





资讯
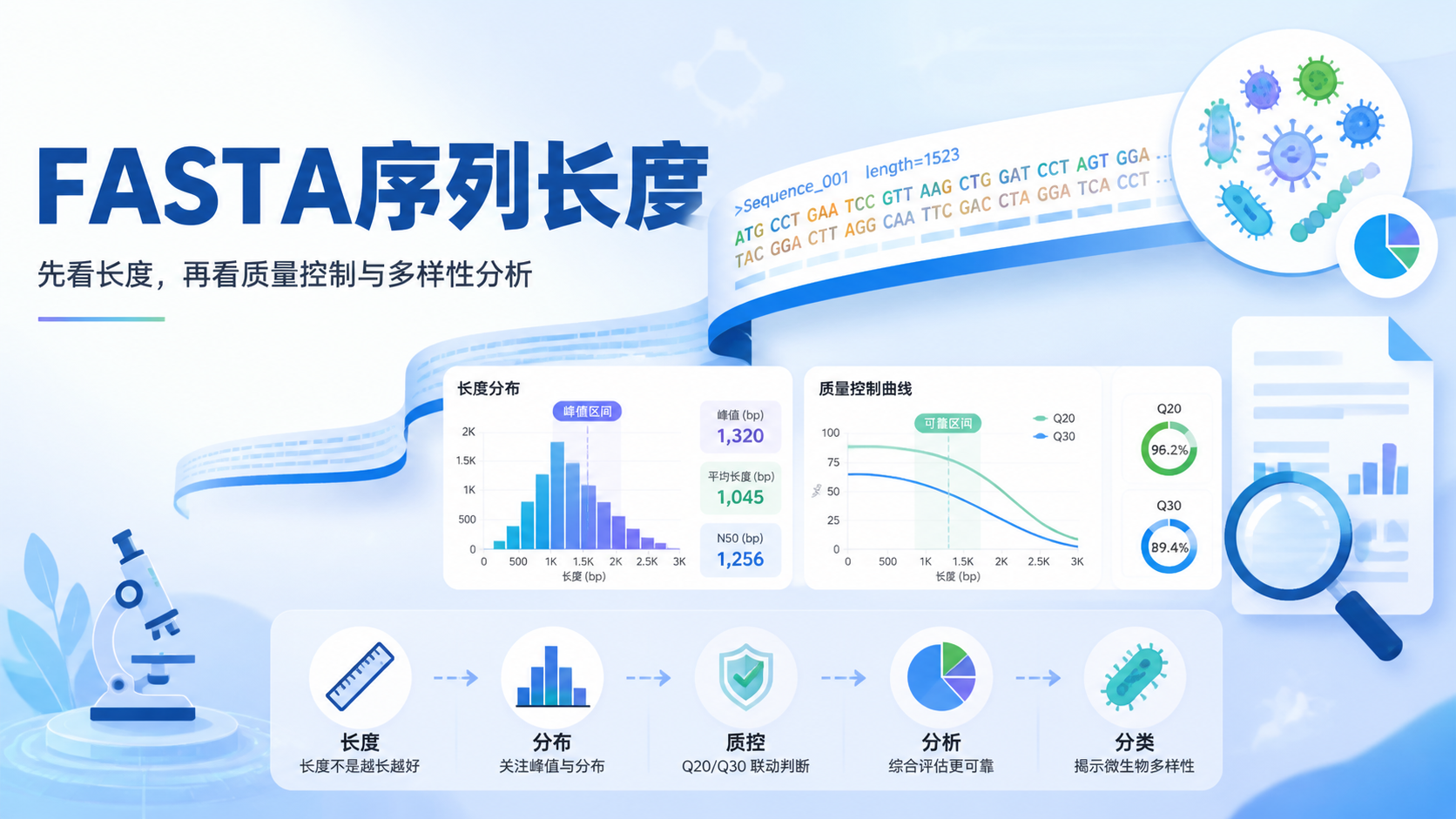 FASTA序列长度看似只是一个数字,却直接影响比对质量、物种注释、OTU划分和后续统计结论。对医学生、医生和科研人员来说,读懂FASTA序列长度,是避免“数据看起来正常,结果却不可靠”的第一步。阅读2 · 点赞0Dr.Sheng 于2026-05-14发布
FASTA序列长度看似只是一个数字,却直接影响比对质量、物种注释、OTU划分和后续统计结论。对医学生、医生和科研人员来说,读懂FASTA序列长度,是避免“数据看起来正常,结果却不可靠”的第一步。阅读2 · 点赞0Dr.Sheng 于2026-05-14发布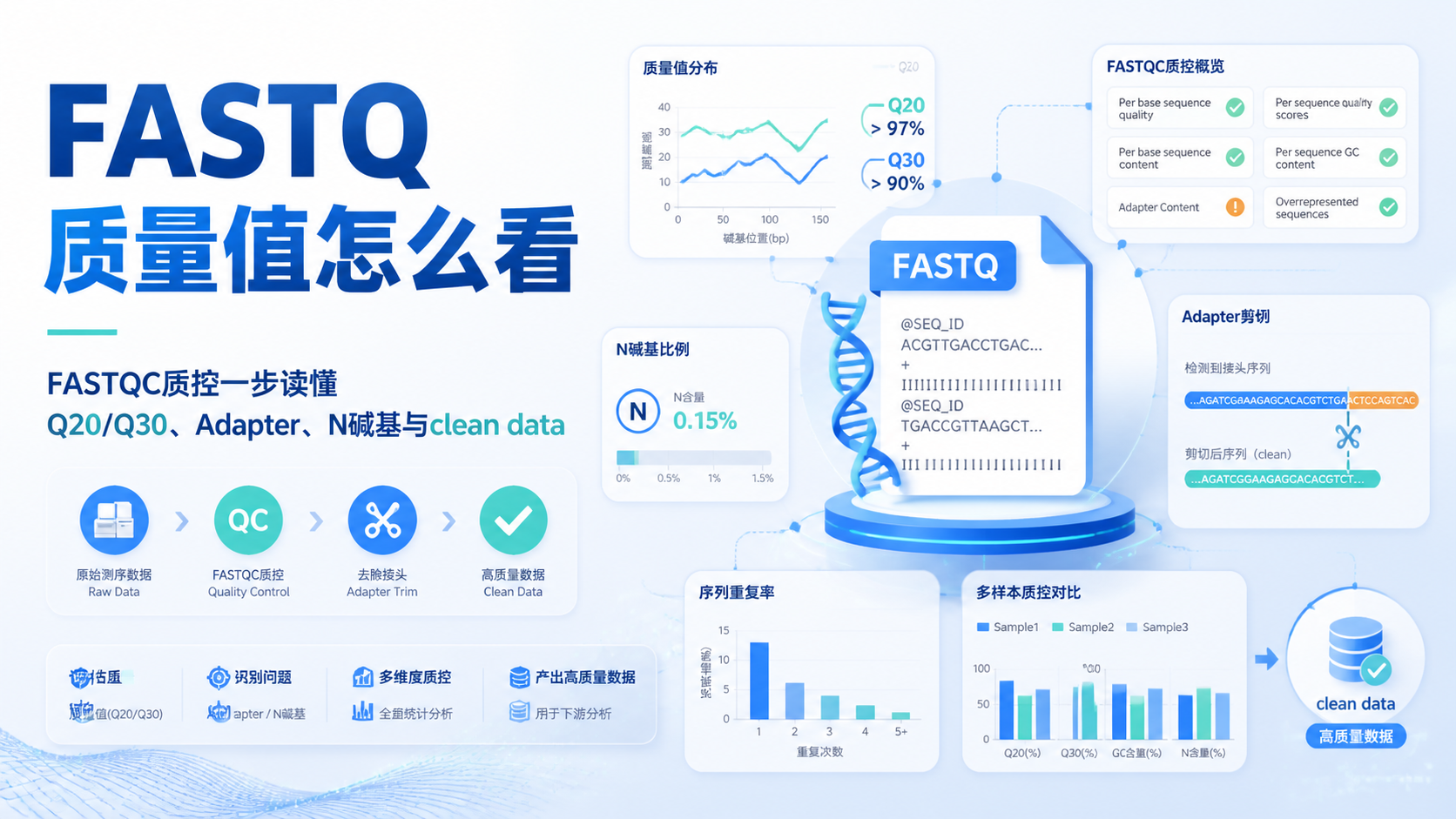
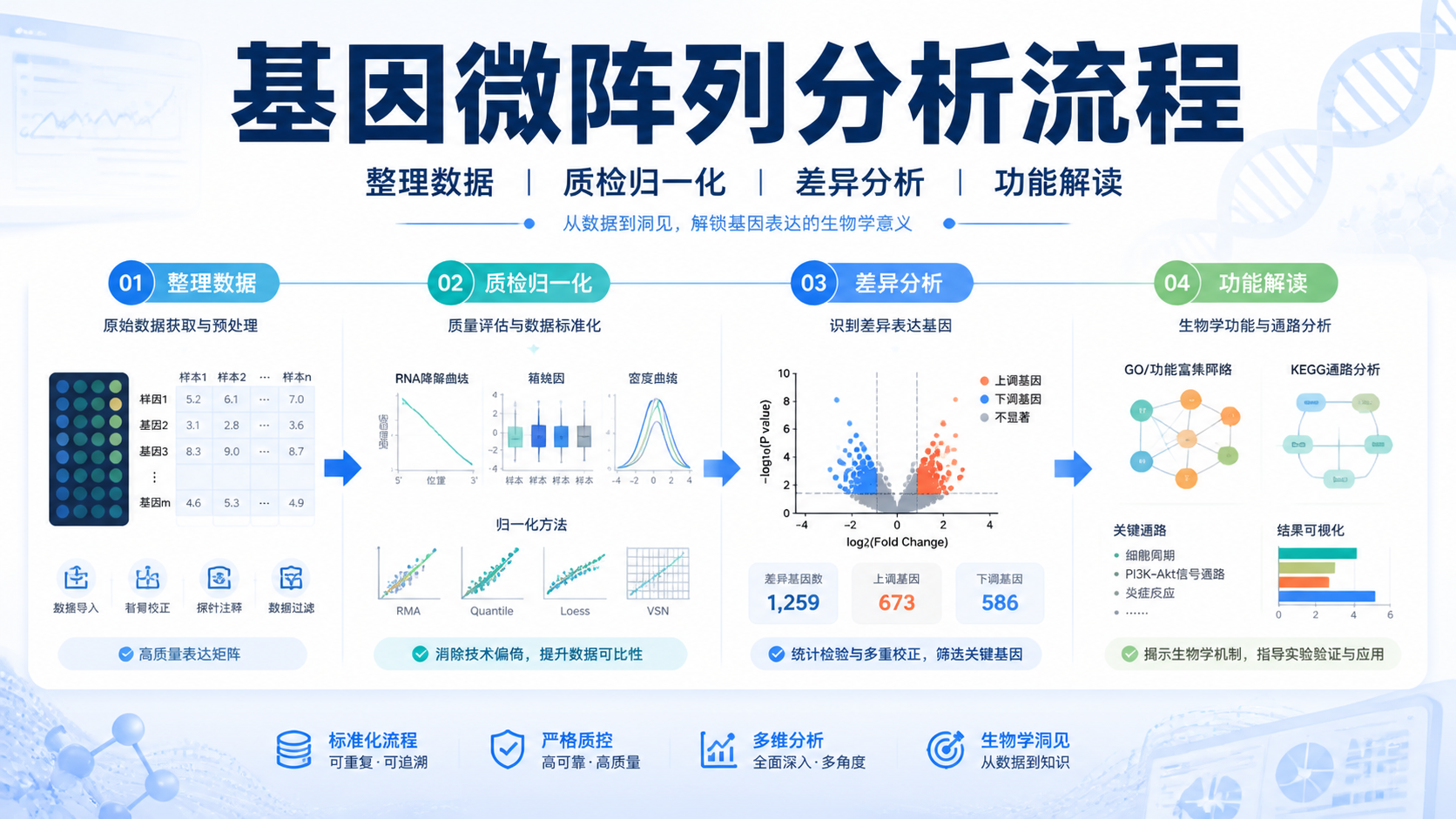 基因微阵列数据怎么做,很多人卡在第一步。文件格式不对、分组不清、归一化混乱,都会让后续差异分析失真。其实,只要把流程拆成3步,就能更稳地完成分析。阅读1 · 点赞0Dr.Sheng 于2026-05-14发布
基因微阵列数据怎么做,很多人卡在第一步。文件格式不对、分组不清、归一化混乱,都会让后续差异分析失真。其实,只要把流程拆成3步,就能更稳地完成分析。阅读1 · 点赞0Dr.Sheng 于2026-05-14发布
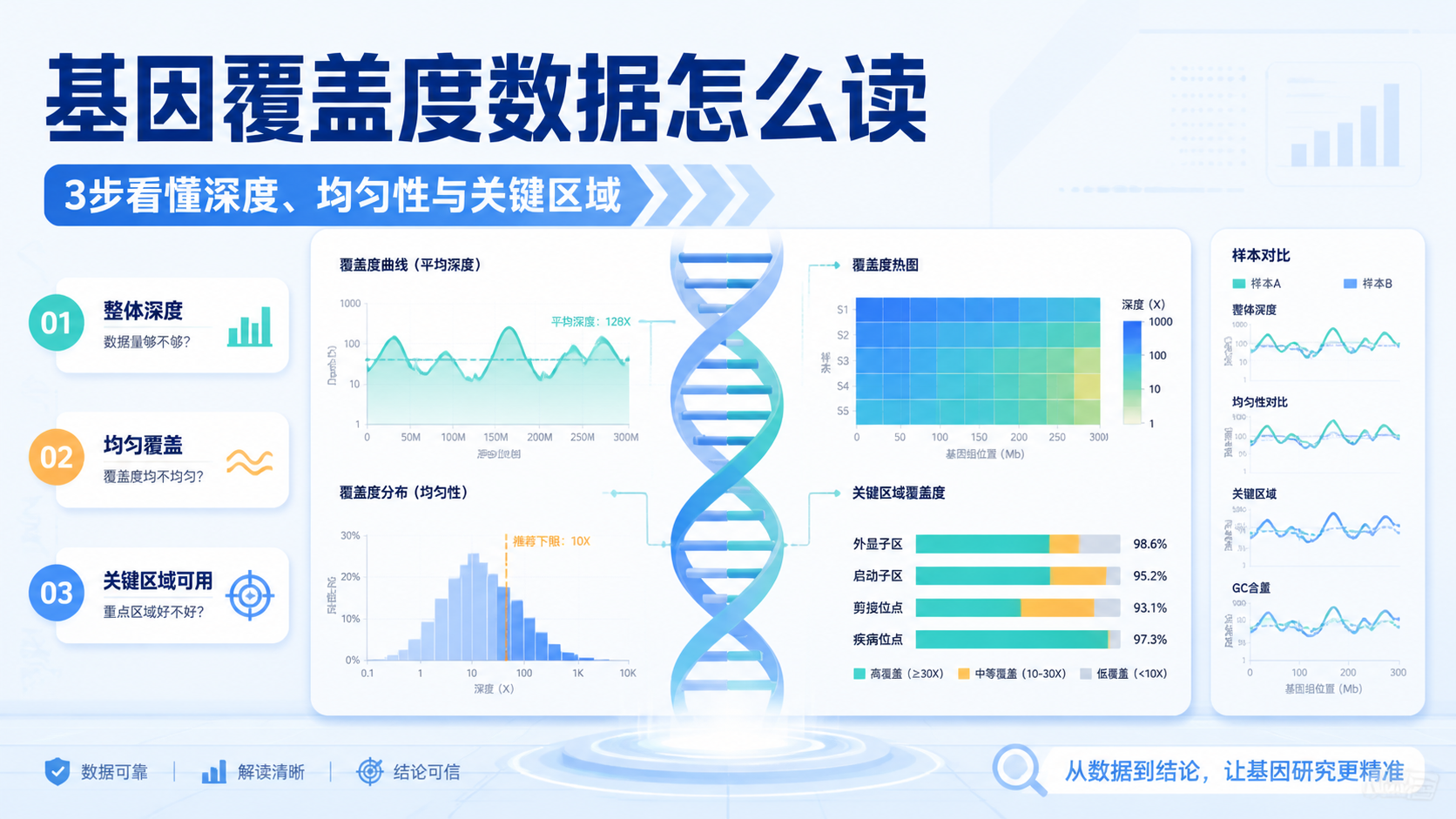 基因覆盖度数据是很多医学生和科研人员在做测序分析时最先遇到、也最容易误读的指标。它直接影响变异检测、拷贝数分析和样本质量判断。如果覆盖度不清楚,后续结果再漂亮也可能不可靠。阅读1 · 点赞0Dr.Sheng 于2026-05-14发布
基因覆盖度数据是很多医学生和科研人员在做测序分析时最先遇到、也最容易误读的指标。它直接影响变异检测、拷贝数分析和样本质量判断。如果覆盖度不清楚,后续结果再漂亮也可能不可靠。阅读1 · 点赞0Dr.Sheng 于2026-05-14发布 测序覆盖度数据直接决定你能否“看见”真实变异。覆盖不够,漏检、假阴性和结果不稳定就会增加。对医学生、医生和科研人员来说,理解测序覆盖度数据,是解读WES、TMB和变异验证结果的基础。阅读1 · 点赞0Dr.Sheng 于2026-05-14发布
测序覆盖度数据直接决定你能否“看见”真实变异。覆盖不够,漏检、假阴性和结果不稳定就会增加。对医学生、医生和科研人员来说,理解测序覆盖度数据,是解读WES、TMB和变异验证结果的基础。阅读1 · 点赞0Dr.Sheng 于2026-05-14发布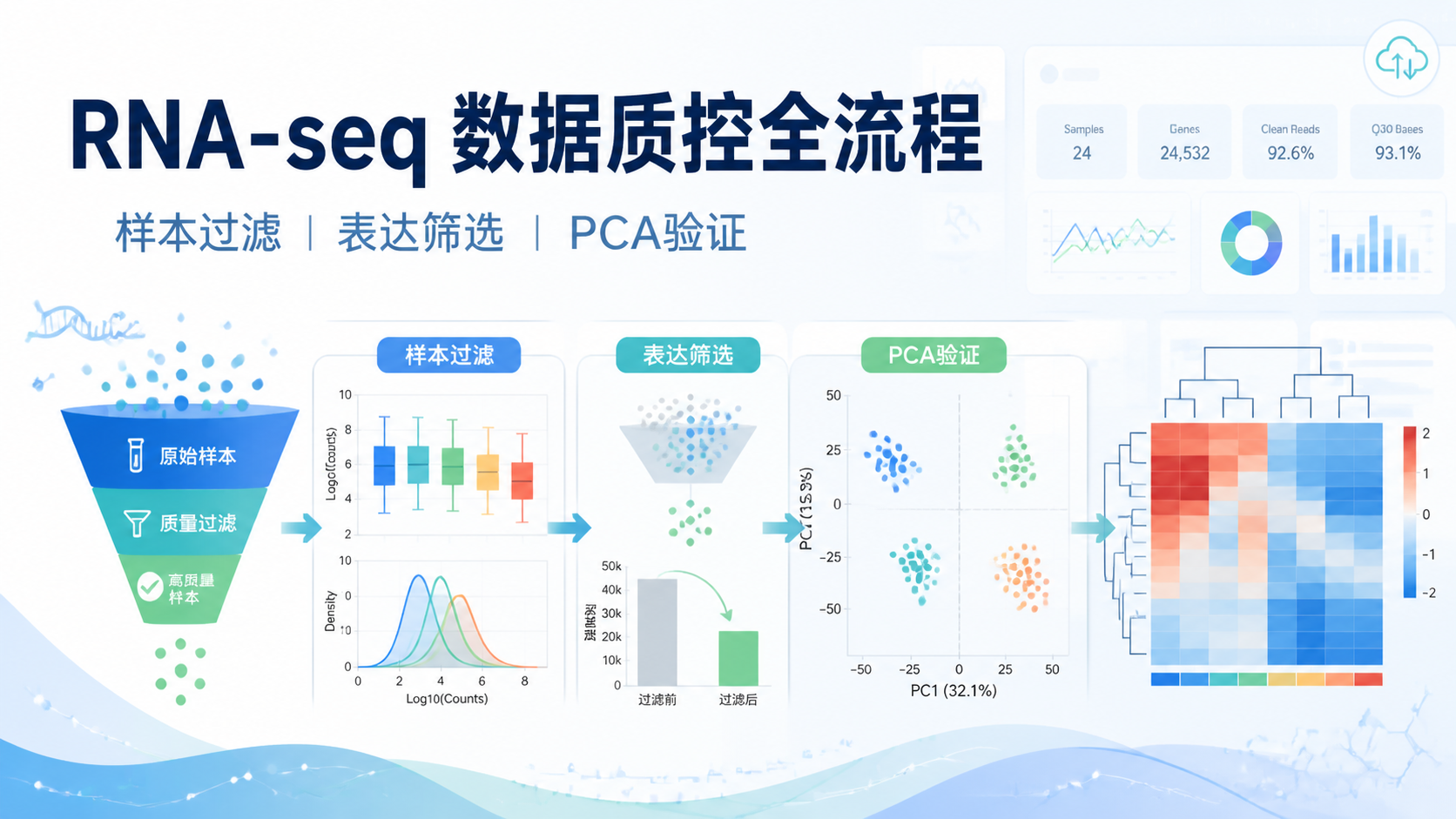 测序深度数据评估,决定了下游分析是否可靠。深度不够,低表达基因会漏检。深度过高但质量差,也会放大噪音。对于医学生、医生和科研人员来说,先看清样本质量,再谈差异分析,才是标准流程。阅读2 · 点赞0Dr.Sheng 于2026-05-14发布
测序深度数据评估,决定了下游分析是否可靠。深度不够,低表达基因会漏检。深度过高但质量差,也会放大噪音。对于医学生、医生和科研人员来说,先看清样本质量,再谈差异分析,才是标准流程。阅读2 · 点赞0Dr.Sheng 于2026-05-14发布 生信索引文件是很多生信分析流程的起点。建错了,后面的比对、注释、检索都会出问题。对医学生、医生和科研人员来说,真正的难点不在“会不会点软件”,而在于是否理解索引文件的用途、结构和构建逻辑。阅读2 · 点赞0Dr.Sheng 于2026-05-14发布
生信索引文件是很多生信分析流程的起点。建错了,后面的比对、注释、检索都会出问题。对医学生、医生和科研人员来说,真正的难点不在“会不会点软件”,而在于是否理解索引文件的用途、结构和构建逻辑。阅读2 · 点赞0Dr.Sheng 于2026-05-14发布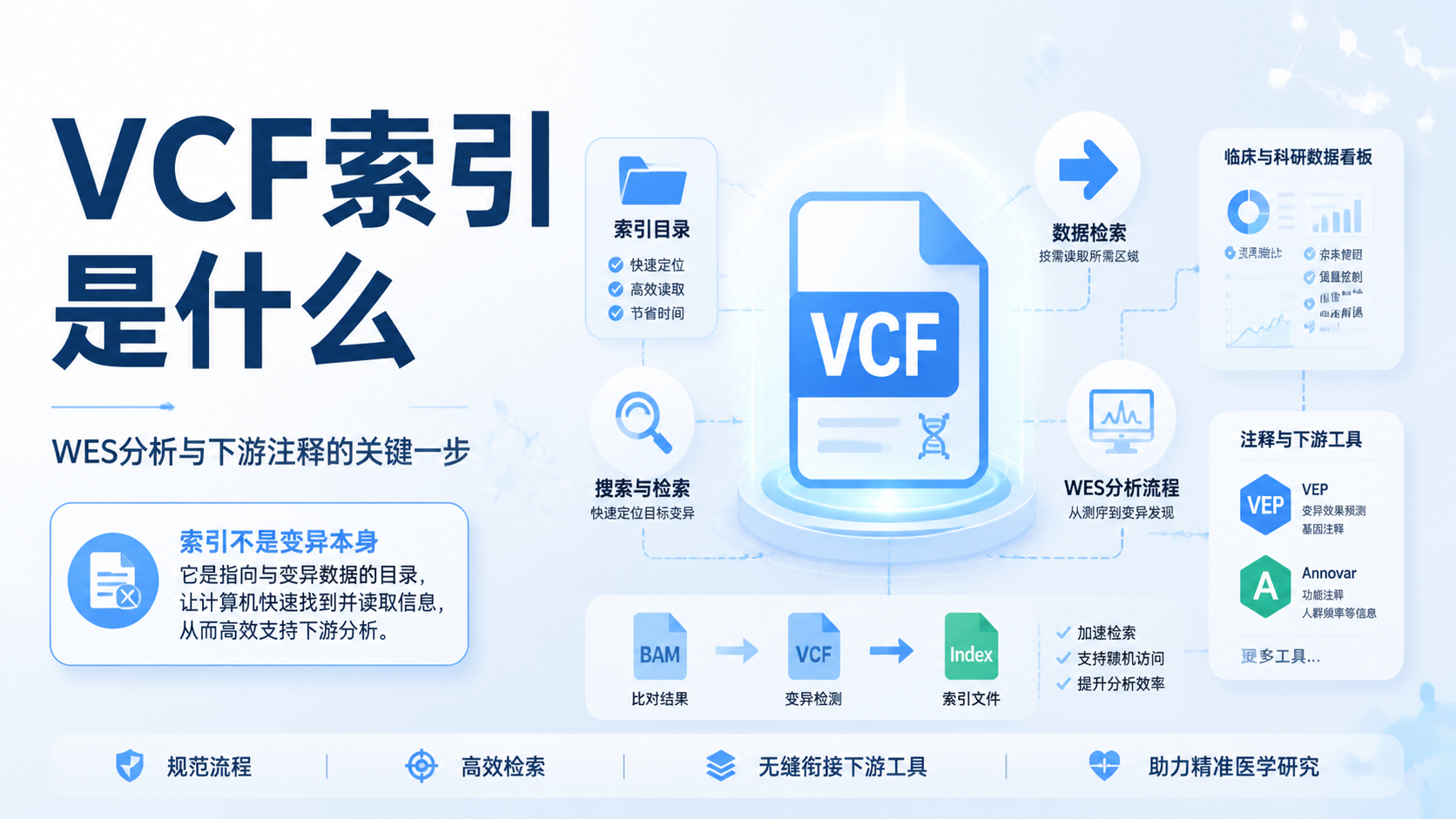 VCF索引是做WES、肿瘤变异分析和临床注释时绕不开的一步。没有索引,VCF文件很难被快速查询、分区读取,也不利于后续质控和自动化流程。对医学生、医生和科研人员来说,理解VCF索引,就是理解变异数据如何高效进入分析和报告环节。阅读2 · 点赞0Dr.Sheng 于2026-05-14发布
VCF索引是做WES、肿瘤变异分析和临床注释时绕不开的一步。没有索引,VCF文件很难被快速查询、分区读取,也不利于后续质控和自动化流程。对医学生、医生和科研人员来说,理解VCF索引,就是理解变异数据如何高效进入分析和报告环节。阅读2 · 点赞0Dr.Sheng 于2026-05-14发布 FASTA索引是测序数据分析的第一步。很多医学生和科研人员拿到序列文件后,常会卡在“怎么建索引、索引为什么失效、下游分析为何报错”。搞懂FASTA索引,才能让比对、检索和注释更稳定。阅读2 · 点赞0Dr.Sheng 于2026-05-14发布
FASTA索引是测序数据分析的第一步。很多医学生和科研人员拿到序列文件后,常会卡在“怎么建索引、索引为什么失效、下游分析为何报错”。搞懂FASTA索引,才能让比对、检索和注释更稳定。阅读2 · 点赞0Dr.Sheng 于2026-05-14发布
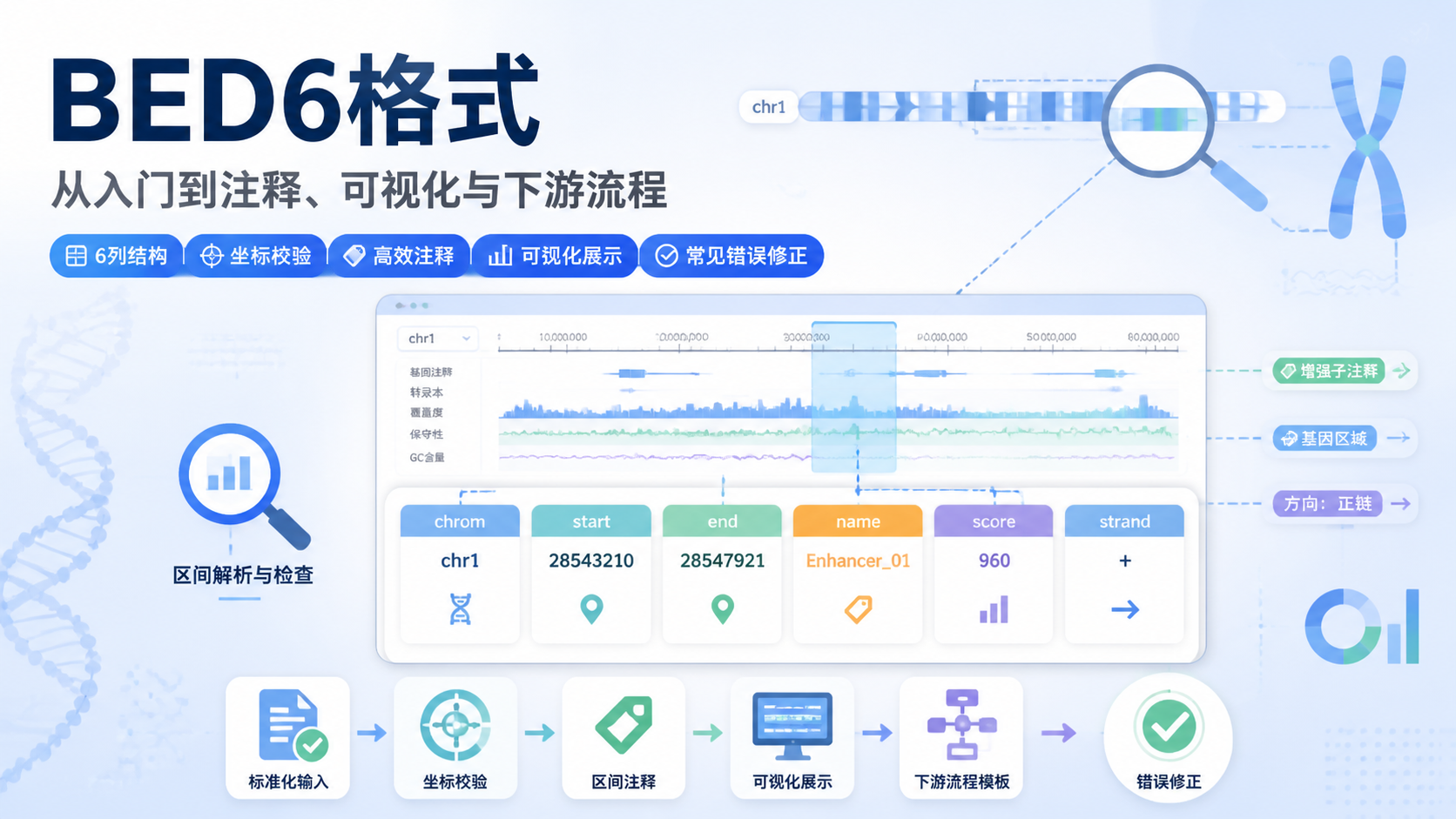
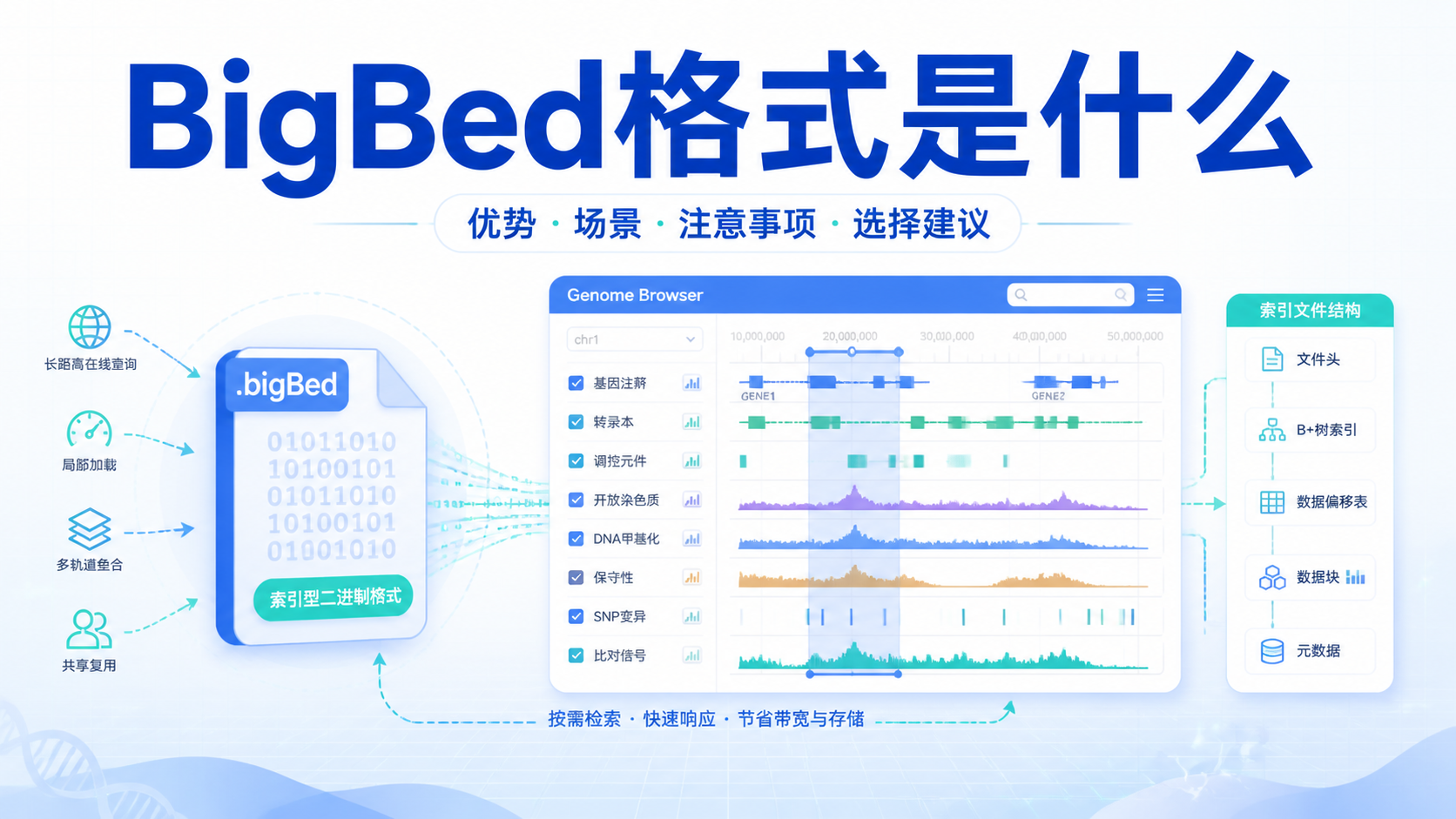
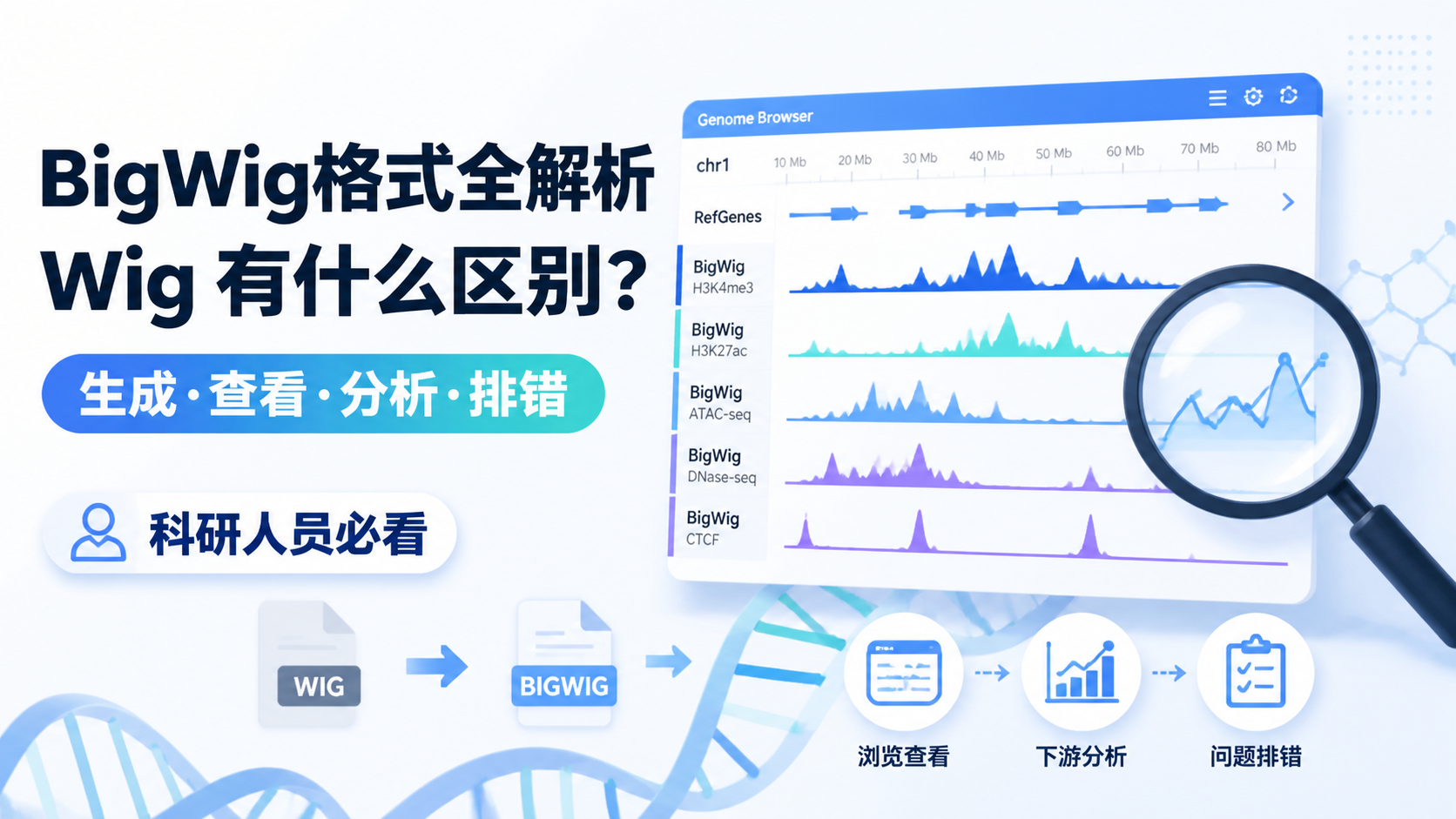


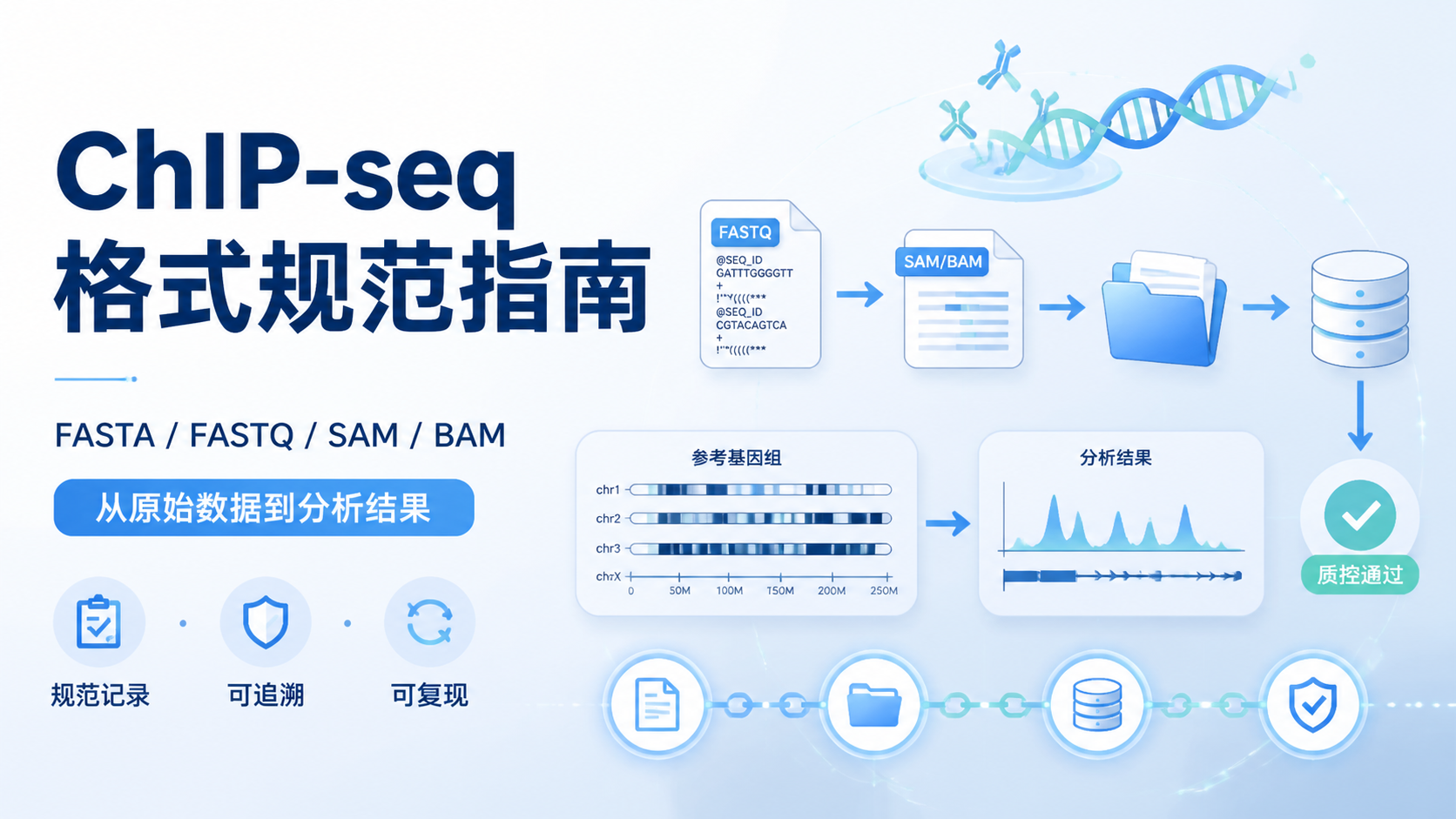 Chip-seq格式直接影响数据存储、比对、质控和下游分析。很多人拿到原始测序文件后,常被FASTA、FASTQ、SAM、BAM等格式绕晕,导致流程衔接不顺、结果难复现。阅读2 · 点赞0Dr.Sheng 于2026-05-14发布
Chip-seq格式直接影响数据存储、比对、质控和下游分析。很多人拿到原始测序文件后,常被FASTA、FASTQ、SAM、BAM等格式绕晕,导致流程衔接不顺、结果难复现。阅读2 · 点赞0Dr.Sheng 于2026-05-14发布 甲基化数据格式看似只是文件整理问题,实际却直接影响比对、注释和下游统计结果。对医学生、医生和科研人员来说,格式选错,后面的差异分析、分组比较和临床关联都可能出偏差。本文围绕甲基化数据格式,拆解最常用的三大核心规范。阅读2 · 点赞0Dr.Sheng 于2026-05-14发布
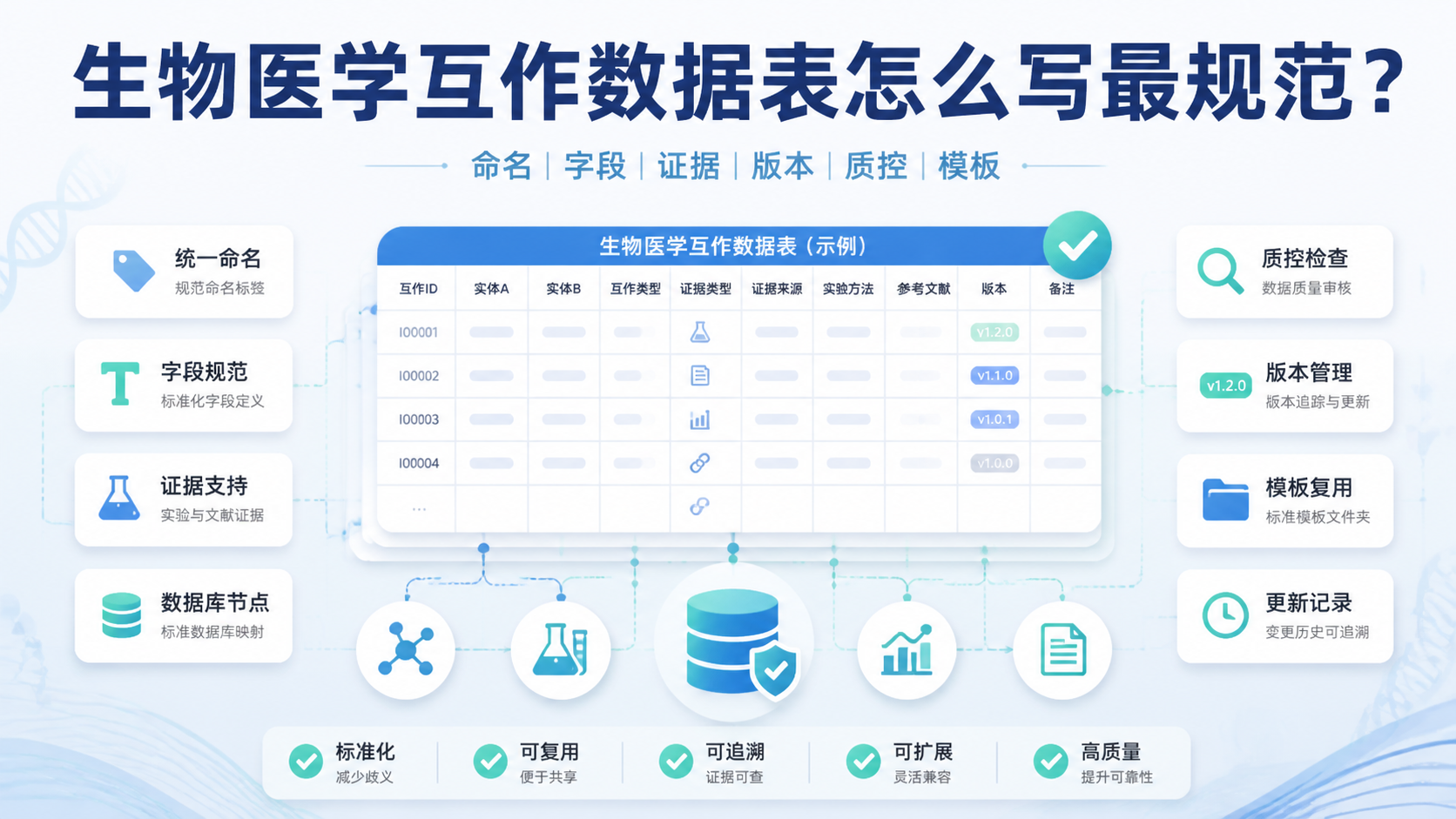
甲基化数据格式看似只是文件整理问题,实际却直接影响比对、注释和下游统计结果。对医学生、医生和科研人员来说,格式选错,后面的差异分析、分组比较和临床关联都可能出偏差。本文围绕甲基化数据格式,拆解最常用的三大核心规范。阅读2 · 点赞0Dr.Sheng 于2026-05-14发布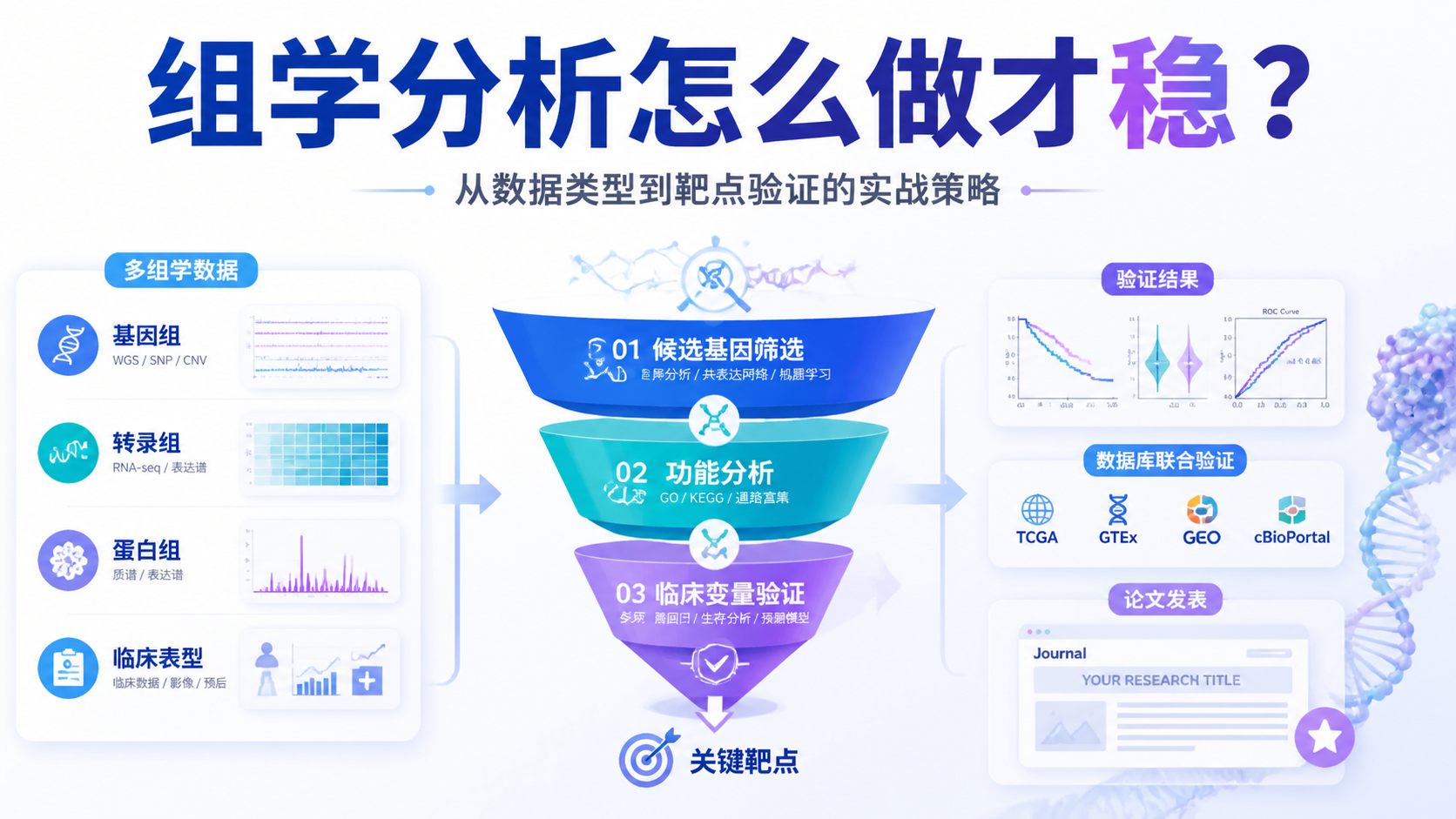 基因组变异数据分析常见痛点,是数据量大、变量多、结果难复现。如果只做单一差异分析,很容易停留在“有结果,但不够强”的层面。本文按生信课题设计思路,拆解3大核心策略,帮助医学生、医生和科研人员更快建立分析框架。阅读5 · 点赞0Dr.Xin 于2026-05-13发布
基因组变异数据分析常见痛点,是数据量大、变量多、结果难复现。如果只做单一差异分析,很容易停留在“有结果,但不够强”的层面。本文按生信课题设计思路,拆解3大核心策略,帮助医学生、医生和科研人员更快建立分析框架。阅读5 · 点赞0Dr.Xin 于2026-05-13发布
热门文章
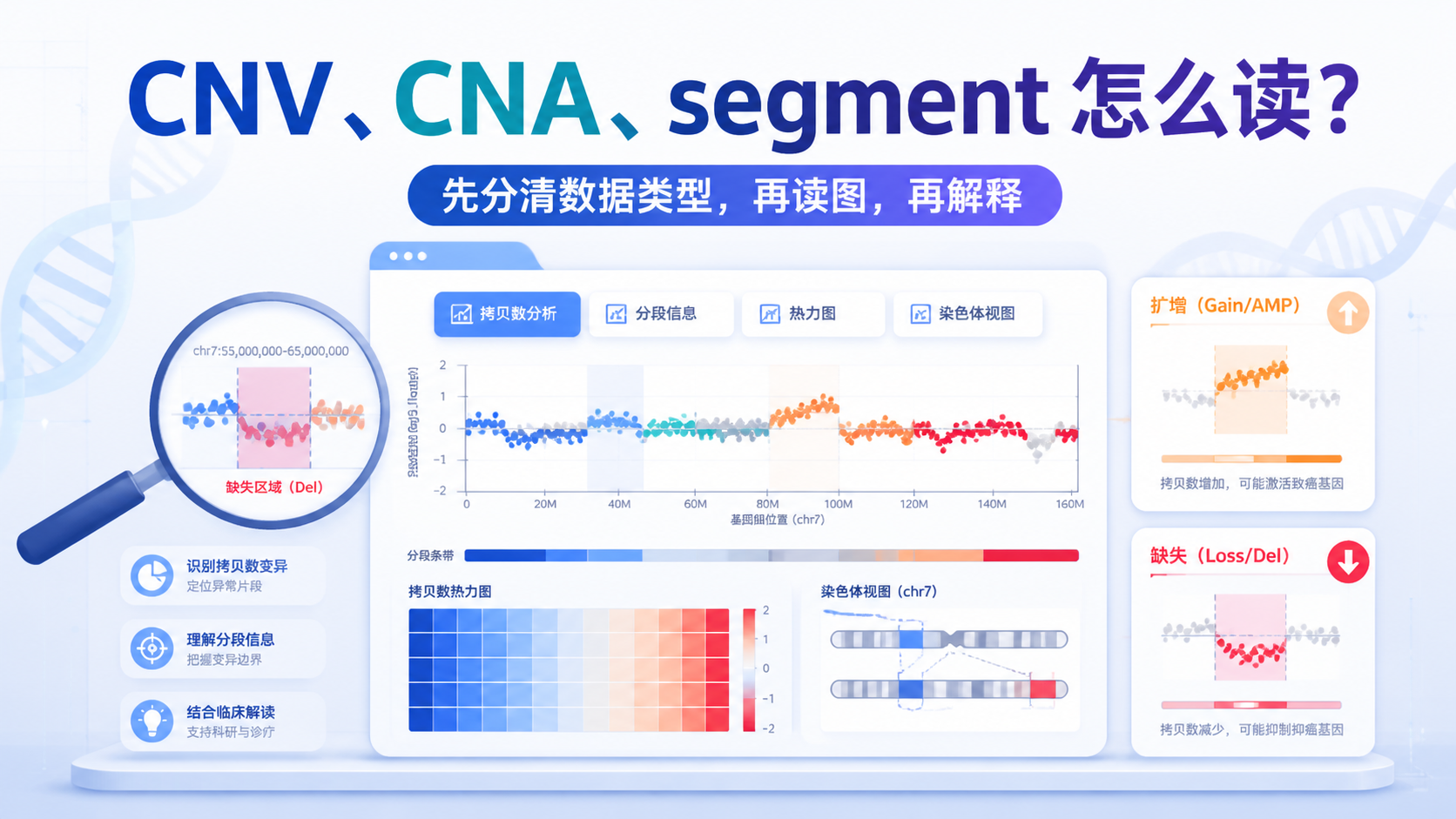
拷贝数变异数据如何解读?3步讲清
阅读40 · 点赞0
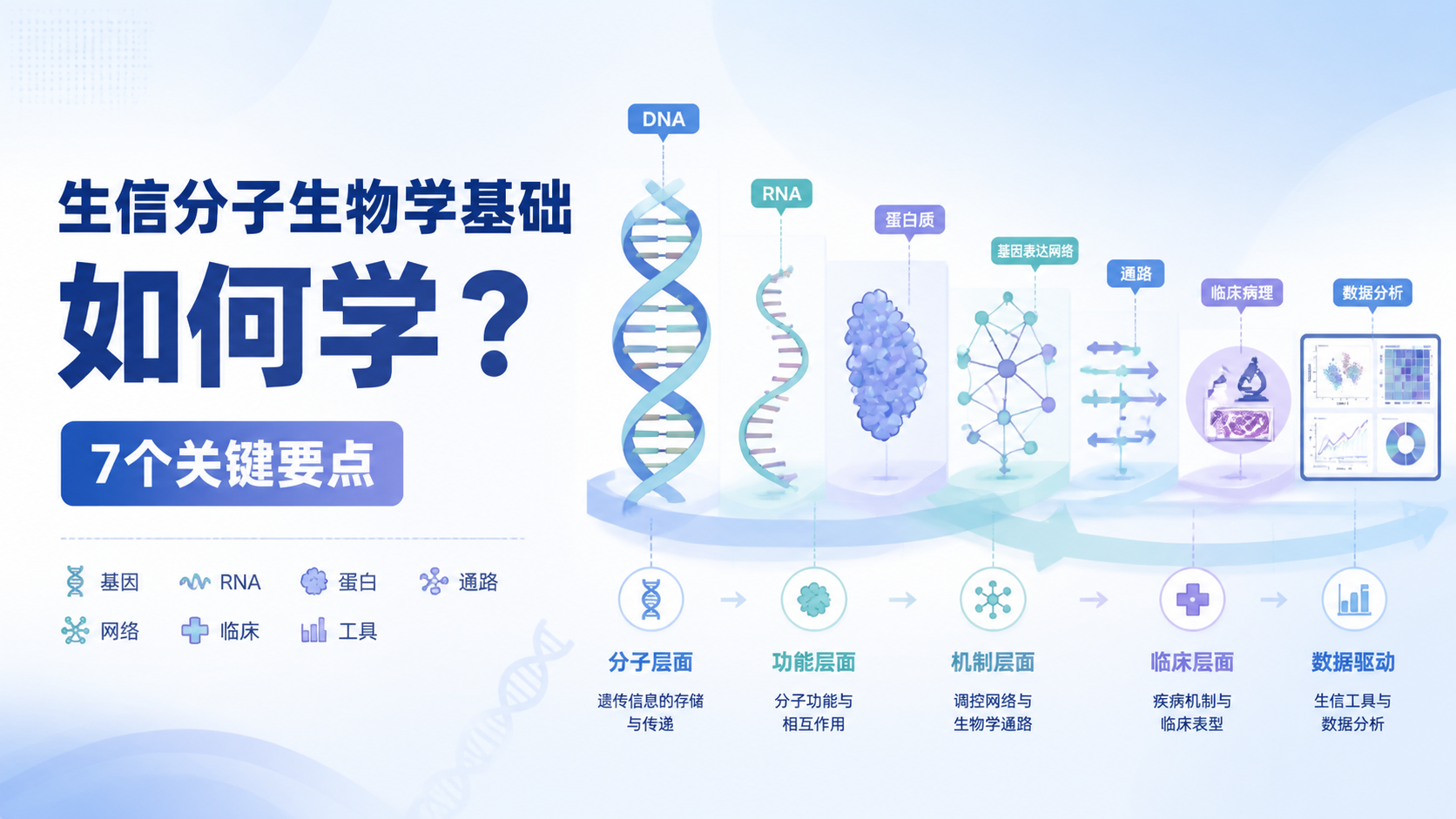
生信遗传学基础怎么学?6个高效方法
阅读29 · 点赞0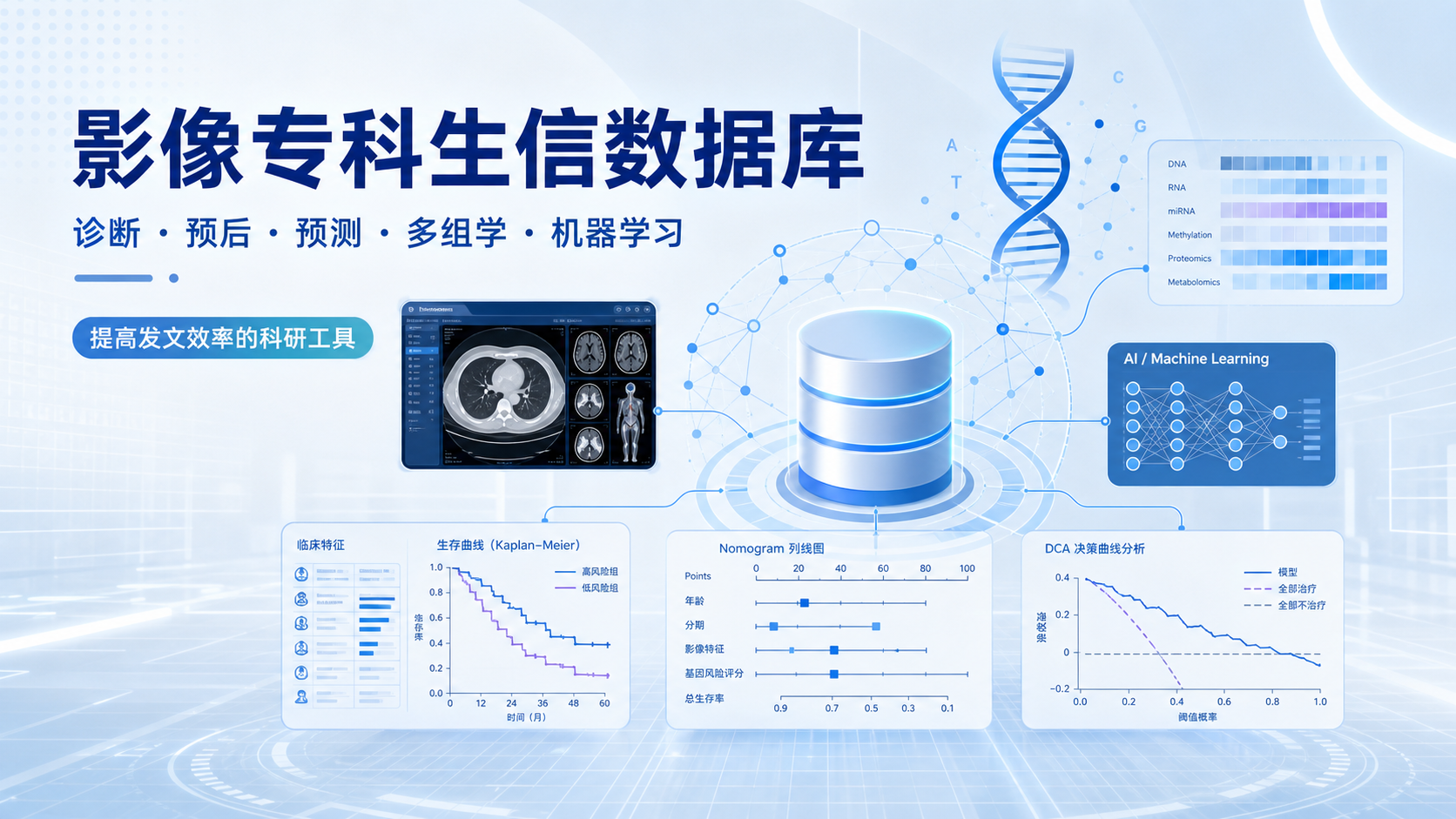
影像专科生信数据库:7个关键应用
阅读26 · 点赞0