引言Introduction
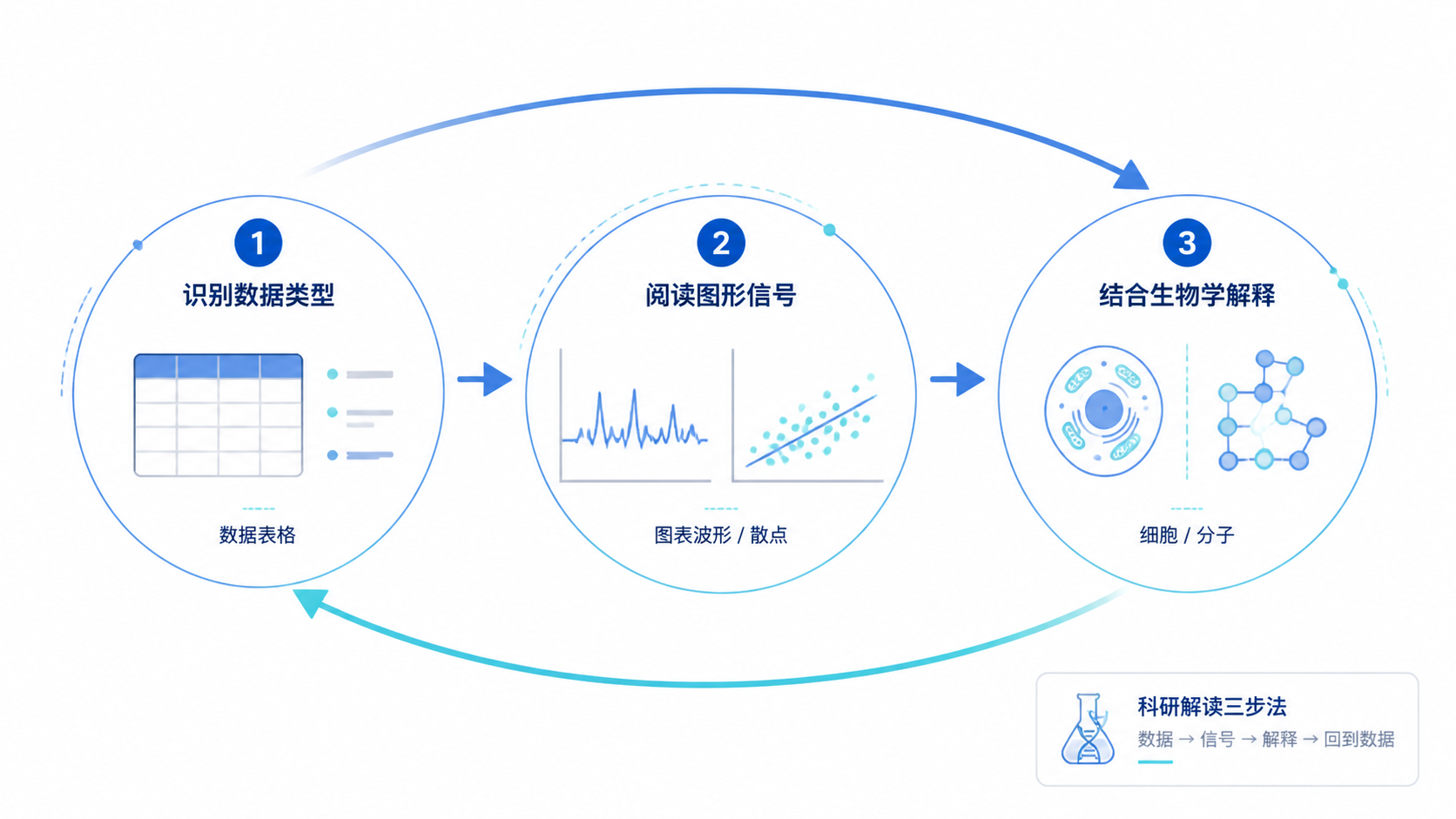
拷贝数变异数据常让人困惑。是扩增,还是缺失。是局部事件,还是大片段改变。对医学生、医生和科研人员来说,关键不是“看见图”,而是读懂信号、判断生物学意义、避免误判。拷贝数变异数据的解读,核心可以拆成3步。
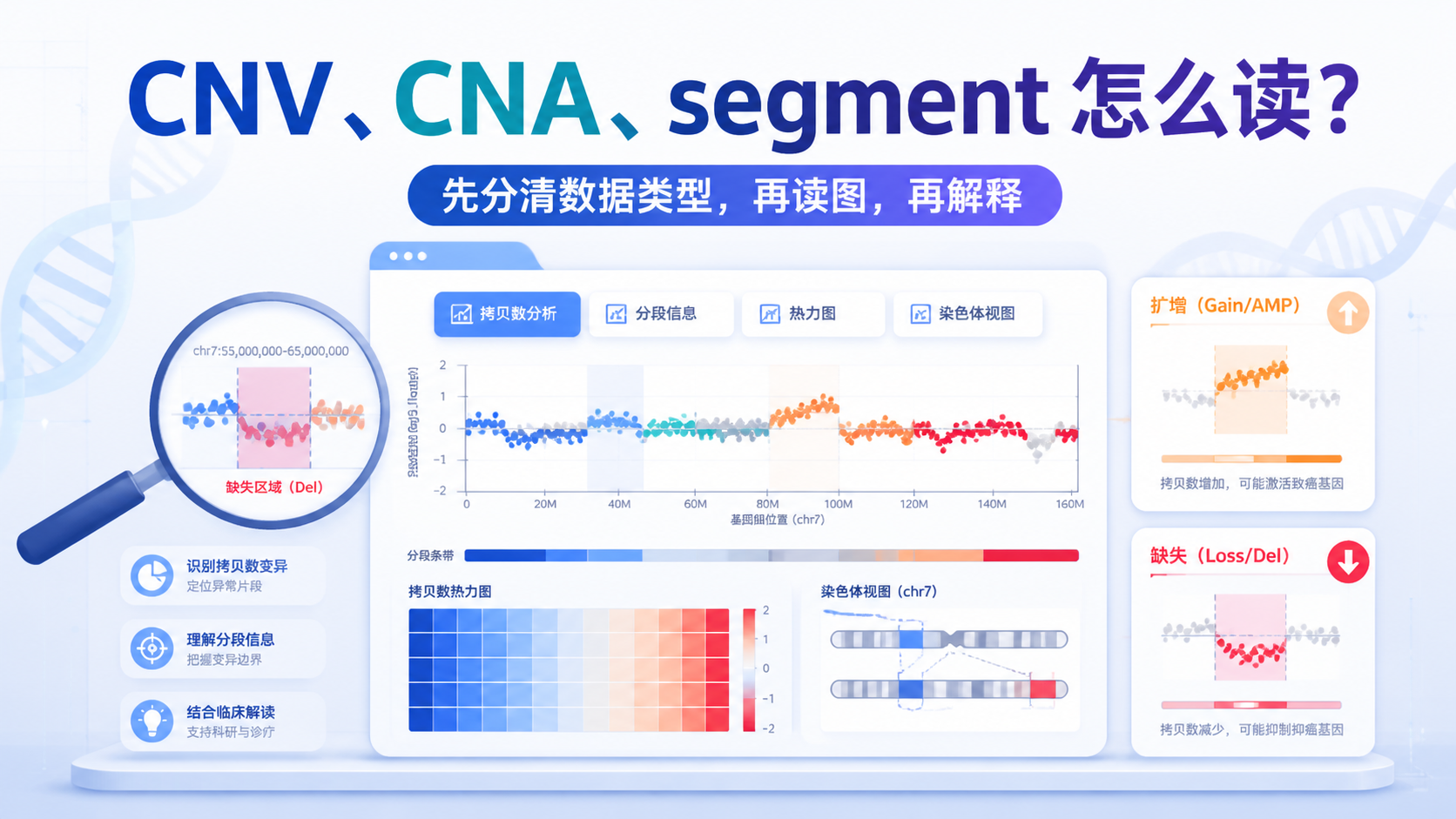
1. 先分清数据类型:你看到的是CNV、CNA还是segment
1.1 数据来源决定解读边界
拷贝数变异数据通常来自测序深度、芯片或分段分析。常见结果包括CNV、CNA和segment文件。它们不是同一层级的数据。
segment文件更接近原始分析结果。 它通常包含样本名、染色体编号、起止位置、片段长度、探针数和平均值。后续很多可视化和统计,都是基于这个层级展开。
1.2 先确认参考基线
解读时一定要先问:参考拷贝数是多少。很多流程里,2通常被定义为正常拷贝数基线 。高于2,多数情况下提示扩增。低于2,多数情况下提示缺失。
但这只是基础规则。不同平台、不同算法、不同纯度样本,阈值可能不同。不先确认基线,后面的扩增和缺失判断都可能偏差。
1.3 关注区间大小和事件类型
拷贝数变异一般指较小尺度的结构性改变。常见定义里,它往往落在1 KB到1 MB之间。更大的改变可能更接近结构变异或大片段染色体异常。
所以解读时要区分:
- 局部基因层面的改变
- 臂水平改变
- 整条染色体异常
这一步的目的很简单。先把数据类型和尺度弄清楚,再谈生物学意义。
2. 再读图:看扩增、缺失和分布模式
2.1 从颜色和方向识别事件
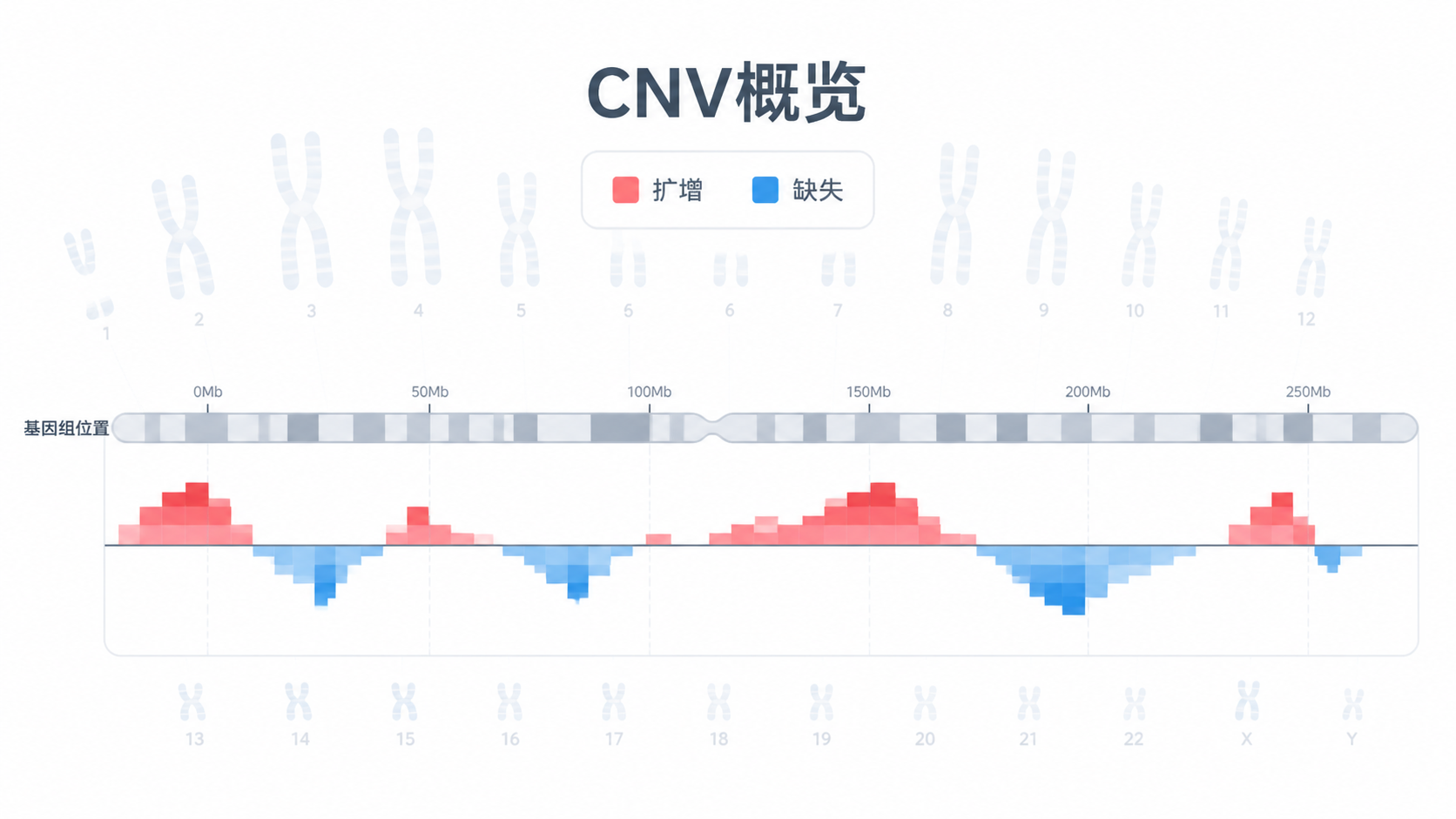
在常见拷贝数可视化中,红色多表示扩增,蓝色多表示缺失。某些图还会用绿色表示缺失。不要只看颜色,要结合图例。
读图时最重要的是看偏离基线的方向。 高于基线是扩增,低于基线是缺失。这个判断在多数CNV分析图里都是第一步。
2.2 判断事件是局部热点还是广泛改变
如果某一段染色体上出现连续偏移,而且集中在较小区域,说明它更像局部拷贝数变异。知识库里的雨点图思路就是这个逻辑。它强调的是事件密集度和位置相关性。
如果某些染色体上出现大片连续改变,比如8号、12号、17号染色体上突变或变异更密集,就提示该区域可能存在更强的聚集效应。局部聚集,往往比零散单点更值得优先关注。
2.3 看样本层面的异质性
同一批样本中,拷贝数变异可能差异很大。部分样本只有少量事件,部分样本则出现明显的广泛改变。解读时要看:
- 有多少样本发生变异
- 哪些基因反复出现
- 是否集中在少数高频区域
如果一个区域在很多样本中重复出现,临床和科研价值通常更高。高频重复事件,优先级通常高于低频偶发事件。
2.4 留意图形展示的默认设置
很多图默认只显示前几个基因名,或自动删除没有变异的样本。这样做是为了提升可读性,但也可能影响解读。
例如,某些绘图函数会把拷贝数为0的样本自动剔除。这样会让总样本数看起来变少。因此,读图前要先确认是否启用了过滤参数。 否则你看到的不是全量数据,而是被筛选后的子集。
3. 最后做解释:把图转成可用结论
3.1 先看是否具有统计和生物学一致性
单个拷贝数变异点不一定有意义。更可靠的信号通常同时满足两个条件:
- 在多个样本中重复出现。
- 落在已知相关基因或功能区域附近。
如果某一区域频繁扩增,且涉及肿瘤驱动相关基因,那么它更可能是值得跟踪的候选位点。统计重复性和功能相关性,缺一不可。
3.2 结合样本背景判断克隆性
拷贝数变异不只是“有没有”,还要看“是不是主克隆事件”。虽然知识库中对等位基因频率的描述更多用于突变频率,但其逻辑对理解克隆结构同样有参考价值。对纯肿瘤样本而言,克隆事件通常更接近主导群体特征。
因此,在解释拷贝数变异数据时,应结合:
- 肿瘤纯度
- 亚克隆结构
- 事件覆盖范围
- 变异强度
同样是扩增,主克隆扩增与局部亚克隆扩增,临床含义不同。
3.3 把结果落到研究问题上
对科研人员来说,最终不是“图好不好看”,而是“这个结果能回答什么问题”。常见落点包括:
- 肿瘤相关通路是否受影响
- 是否存在特定染色体热点
- 样本间是否存在分型差异
- 拷贝数改变是否与其他组学一致
如果你能把拷贝数变异数据和突变、表达、临床分期一起看,结论会更稳。单一视角容易误读,多组学整合更接近真实生物学。
4. 实操上怎么做,才不容易出错
4.1 先检查文件格式和字段
在实际分析里,最常见的错误不是算法本身,而是输入文件不规范。比如样本名列、染色体列、起止位置列、segment均值列缺失,都会影响绘图和统计。
建议先核对:
- 样本名是否唯一
- 染色体命名是否统一
- 坐标是否为同一版本
- segment平均值是否可比较
4.2 先定阈值,再解释结果
不要看完图再倒推阈值。应先明确扩增和缺失的判定标准,再分析结果。否则不同项目之间无法比较。
同一套阈值应该在同一批样本中保持一致。 这对队列研究尤其重要。
4.3 输出要兼顾可读性和信息量
如果只显示少数基因名,图会更清爽,但细节会减少。如果显示全部基因,又可能遮挡严重。知识库中也提到,文字可能超出边界,需要调整字体大小。
这说明,高质量的拷贝数变异数据展示,不只是统计正确,还要保证信息可读。
总结Conclusion
拷贝数变异数据的解读,关键就是三步。先分清数据类型和基线,再读图识别扩增与缺失,最后把结果放回生物学和临床语境中解释。 只看颜色不够,只看单点也不够。真正有价值的,是能够重复、可验证、可用于研究设计的信号。
如果你希望把拷贝数变异数据快速整理成可分析、可作图、可发表的结果,可以关注解螺旋 。它能帮助你更高效地完成数据整理与可视化,让研究结论更清晰,论文表达更规范。
- 引言Introduction
- 1. 先分清数据类型:你看到的是CNV、CNA还是segment
- 2. 再读图:看扩增、缺失和分布模式
- 3. 最后做解释:把图转成可用结论
- 4. 实操上怎么做,才不容易出错
- 总结Conclusion