引言Introduction
测序比对数据是RNA-seq分析中最容易被忽视,却最影响结论可靠性的环节。很多人拿到比对结果,只看“比对率高不高”,却忽略了数据是否可用、是否均匀、是否存在偏倚。真正读懂测序比对数据,才能判断后续差异分析是否可信。
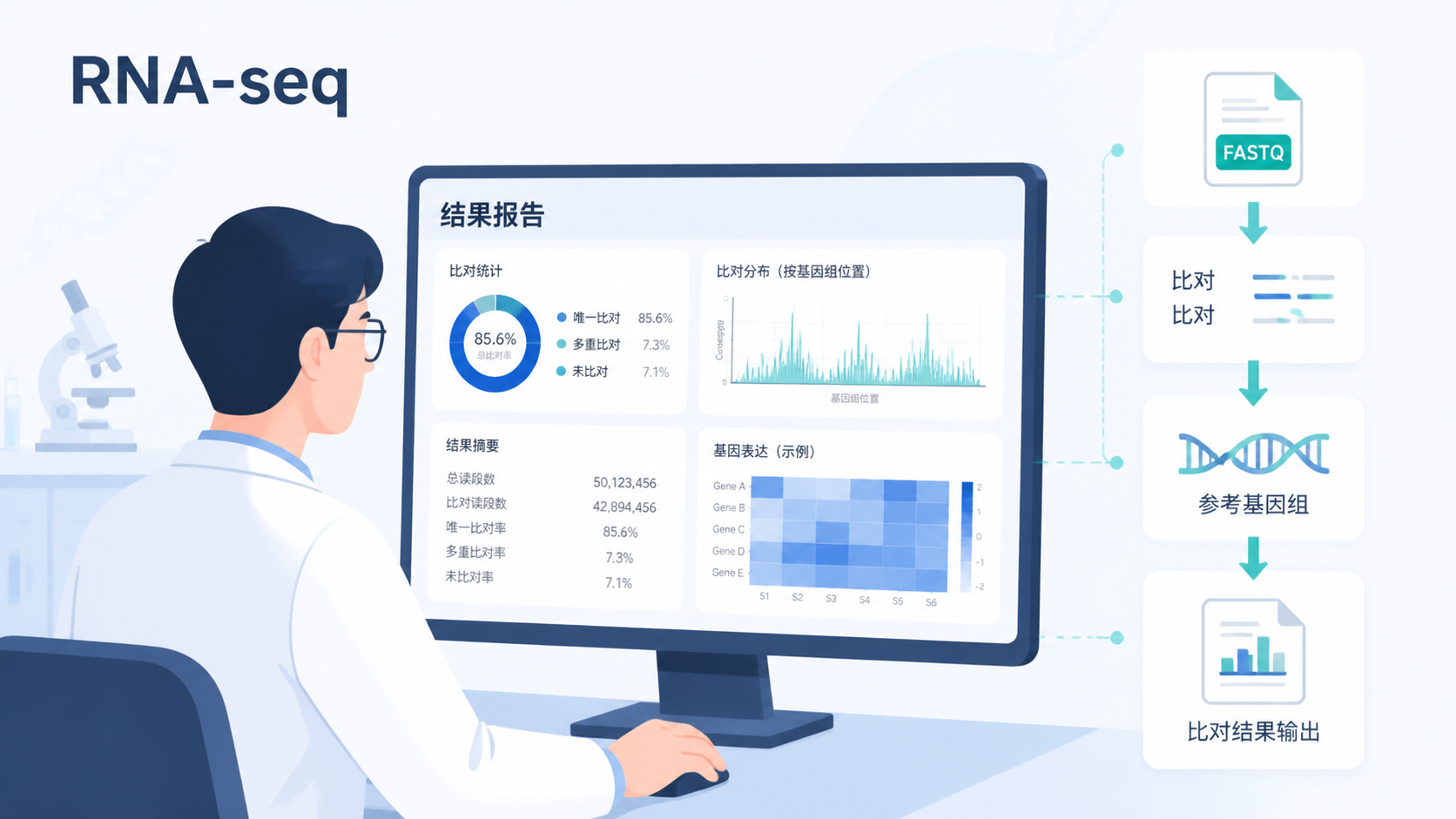
1. 先确认:你的数据是否已经进入可分析状态
1.1 从raw data到clean data,是解读比对数据的前提
在看测序比对数据之前,先要确认原始FASTQ是否经过质控。FASTQ文件每条read通常有4行,第二行是序列,第四行是质量值。Q30代表碱基准确率达到99.9%,这是常用的基础判断标准。
如果质控不过关,后面的比对结果再漂亮也没有意义。因为低质量碱基、接头污染、N值过多,都会直接影响比对率和后续定量。RNA-seq流程里,先做QC和filter,再进入比对,这是基本顺序。
1.2 测序比对数据不是孤立指标
测序比对数据的意义,不只是“对上了多少reads”。 它是连接原始测序和表达矩阵的桥梁。只有比对结果可信,才能继续做表达定量、差异基因分析和功能注释。
对于医学生、医生和科研人员来说,最重要的不是记住软件名,而是理解:比对是把短reads放回参考基因组的过程。这个过程是否顺利,决定了后续结果的可解释性。
2. 第一个关键点:看比对率,但不要只看比对率
2.1 比对率反映了reads能否找到参考序列位置
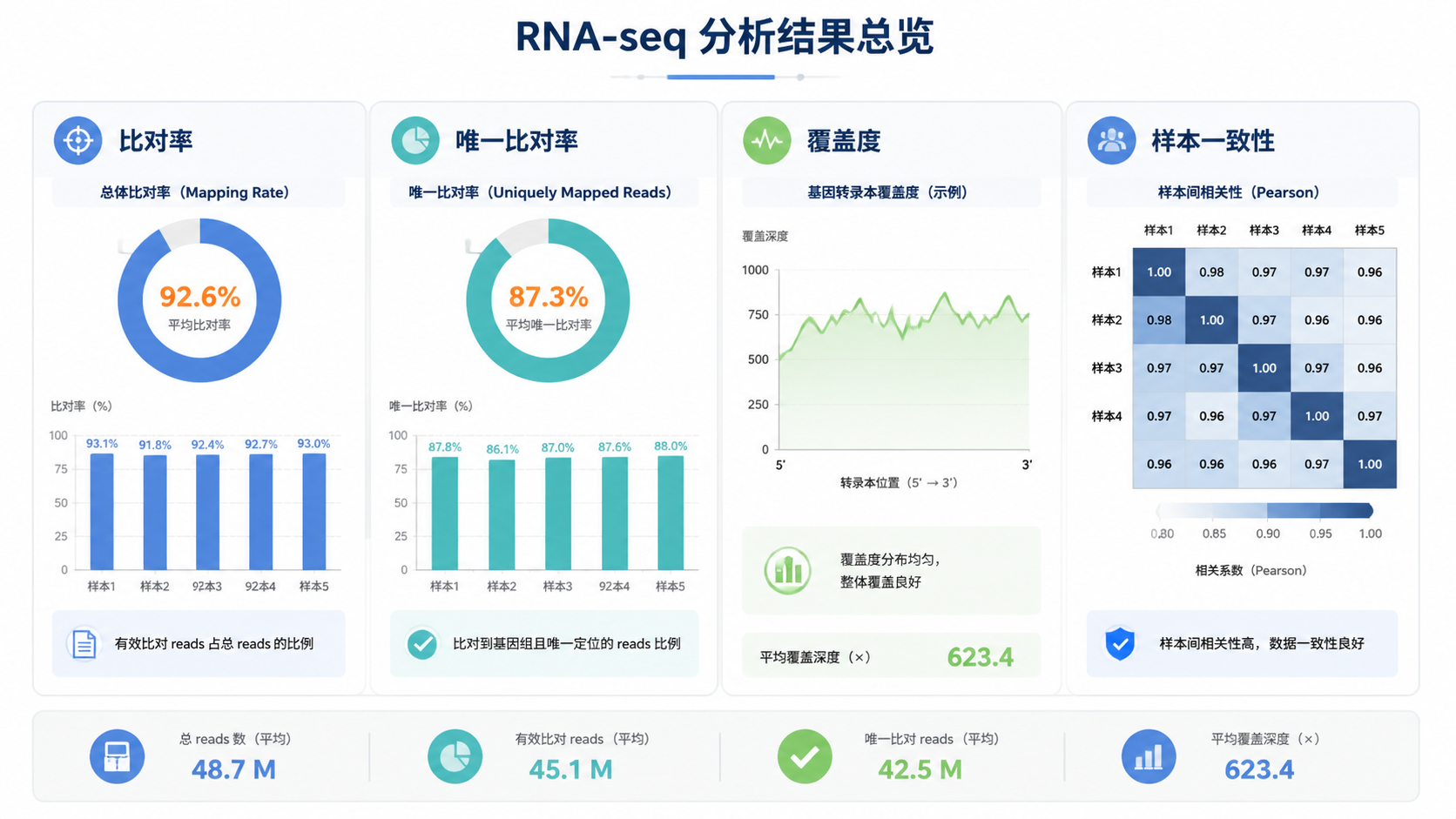
测序比对数据最常见的指标就是比对率。它表示有多少reads成功比对到参考基因组或转录组。比对率高,通常说明样本质量、建库和测序整体情况较好。
但比对率不是唯一标准。不同物种、不同参考库、不同建库方式,都会影响比对结果。比如参考基因组不完整,或者样本污染较多,比对率就可能下降。
2.2 低比对率要先查原因
如果测序比对数据中比对率偏低,常见原因包括:
- 样本RNA降解,导致片段过短或破碎。
- 接头污染或低质量碱基过多。
- 参考基因组版本不匹配。
- 样本中存在非目标物种序列。
- 文库构建质量差。
不要直接把低比对率等同于“数据不能用”。 正确做法是先排查质控、参考库和样本来源,再决定是否重做分析。
3. 第二个关键点:看唯一比对和多重比对
3.1 唯一比对更能代表真实信号
测序比对数据里,一个核心问题是reads到底比到哪里。唯一比对,指一个read只落在基因组的一个位置。 这类reads通常更适合用于下游定量,因为歧义更少。
多重比对则意味着同一个read能比到多个位置。常见于重复序列、同源基因家族,或者基因组中高度相似区域。多重比对过多时,定量结果会受影响。
3.2 读懂“重复”和“歧义”很重要
在RNA-seq中,有些重复并不一定是坏事。因为一个高表达基因本来就会产生大量reads,重复水平偏高并不罕见。真正要警惕的是异常高的非特异性重复和大量无法唯一定位的reads。
如果一个样本的唯一比对比例明显低于同批次其他样本,就要重点检查是否存在污染、参考库不合适,或者建库片段太短。
4. 第三个关键点:看比对到基因组的区域分布
4.1 比对区域能提示样本类型是否合理
测序比对数据不仅要看“比上没比上”,还要看“比到哪里去了”。对于RNA-seq,合理的reads应主要分布在外显子区域。因为成熟mRNA经过剪接后,外显子信号更丰富。
如果大量reads比到内含子或基因间区,可能提示以下问题:
- 样本RNA完整性差。
- 转录本未充分纯化。
- 文库中混入较多基因组DNA。
- 研究对象本身转录活性异常。
4.2 区域分布不对,定量会受影响
比对区域分布异常,往往意味着后续表达定量会偏离真实生物学信号。 尤其是临床样本、低起始量样本和降解样本,更需要关注这一点。
对于科研人员来说,不能只接受“软件跑完了”这个结果。更应问自己:这些reads是否落在我预期的生物学区域内。
5. 第四个关键点:看覆盖度和均匀性
5.1 覆盖是否均匀,决定表达是否稳定
测序比对数据中,覆盖度是另一个关键指标。它反映reads在基因组或转录本上的分布情况。理想状态下,覆盖应该相对均匀。
如果覆盖明显偏向某一端,可能与建库偏倚、RNA降解或扩增偏倚有关。比如3’端偏倚常见于部分降解样本,这会影响全长转录本分析。
5.2 均匀性差,说明数据解释要谨慎
覆盖不均会带来两个问题:
- 低表达基因更容易被漏检。
- 绝对表达量比较会不稳定。
所以在解读测序比对数据时,不要只盯着总reads数。还要看覆盖是否均匀,尤其是在做差异分析、可变剪接分析和融合基因分析前。
6. 第五个关键点:看样本间一致性和批次差异
6.1 同组样本应当具有相近的比对特征
如果同一组样本的测序比对数据差异很大,就要警惕批次效应或样本质量差异。常见表现包括比对率波动大、唯一比对率差异明显、覆盖偏倚不一致。
同组一致性差,会直接削弱统计分析的可信度。 即使后续差异基因很多,也可能只是技术噪音。
6.2 结合实验背景一起判断
解读测序比对数据时,一定要结合实验设计。比如组织样本、血液样本、肿瘤样本、单细胞样本,本身就有不同的复杂度。不同类型数据没有绝对统一阈值,必须结合场景判断。
建议至少检查以下内容:
- 每个样本的总reads数是否接近。
- 比对率是否在同一批次内稳定。
- 唯一比对比例是否异常。
- 覆盖分布是否有明显偏差。
- 是否存在明显离群样本。
7. 如何把测序比对数据用于实际判断
7.1 一个实用的判断顺序
面对测序比对数据,可以按以下顺序看:
- 先看质控是否通过。
- 再看总体比对率。
- 再看唯一比对和多重比对。
- 接着看比对区域与覆盖均匀性。
- 最后看样本间一致性。
这个顺序能帮助你快速判断数据是否值得进入下游分析。
7.2 不要只依赖单一数字
很多初学者喜欢问“比对率多少才算合格”。事实上,单一阈值不能解决所有问题。测序比对数据的解读,本质上是对质量、来源、结构和一致性的综合判断。
对于不同研究场景,判断标准也会不同。临床样本常受降解影响,单细胞样本常受起始量影响,肿瘤样本常受异质性影响。理解这些背景,才能避免误判。
总结Conclusion
测序比对数据的解读,核心不是看一个数字,而是看五个关键点。第一,看是否完成高质量质控。第二,看比对率。第三,看唯一比对和多重比对。第四,看区域分布和覆盖均匀性。第五,看样本间一致性。只有把这些信息放在一起,才能判断数据是否适合进入后续分析。
如果你正在做RNA-seq或相关组学分析,建议把质控和比对结果放在同一条逻辑链里审视。需要更高效地完成数据整理、结果解读和图表输出时,可以关注解螺旋 ,用更系统的工具和内容支持你的科研分析流程。
- 引言Introduction
- 1. 先确认:你的数据是否已经进入可分析状态
- 2. 第一个关键点:看比对率,但不要只看比对率
- 3. 第二个关键点:看唯一比对和多重比对
- 4. 第三个关键点:看比对到基因组的区域分布
- 5. 第四个关键点:看覆盖度和均匀性
- 6. 第五个关键点:看样本间一致性和批次差异
- 7. 如何把测序比对数据用于实际判断
- 总结Conclusion