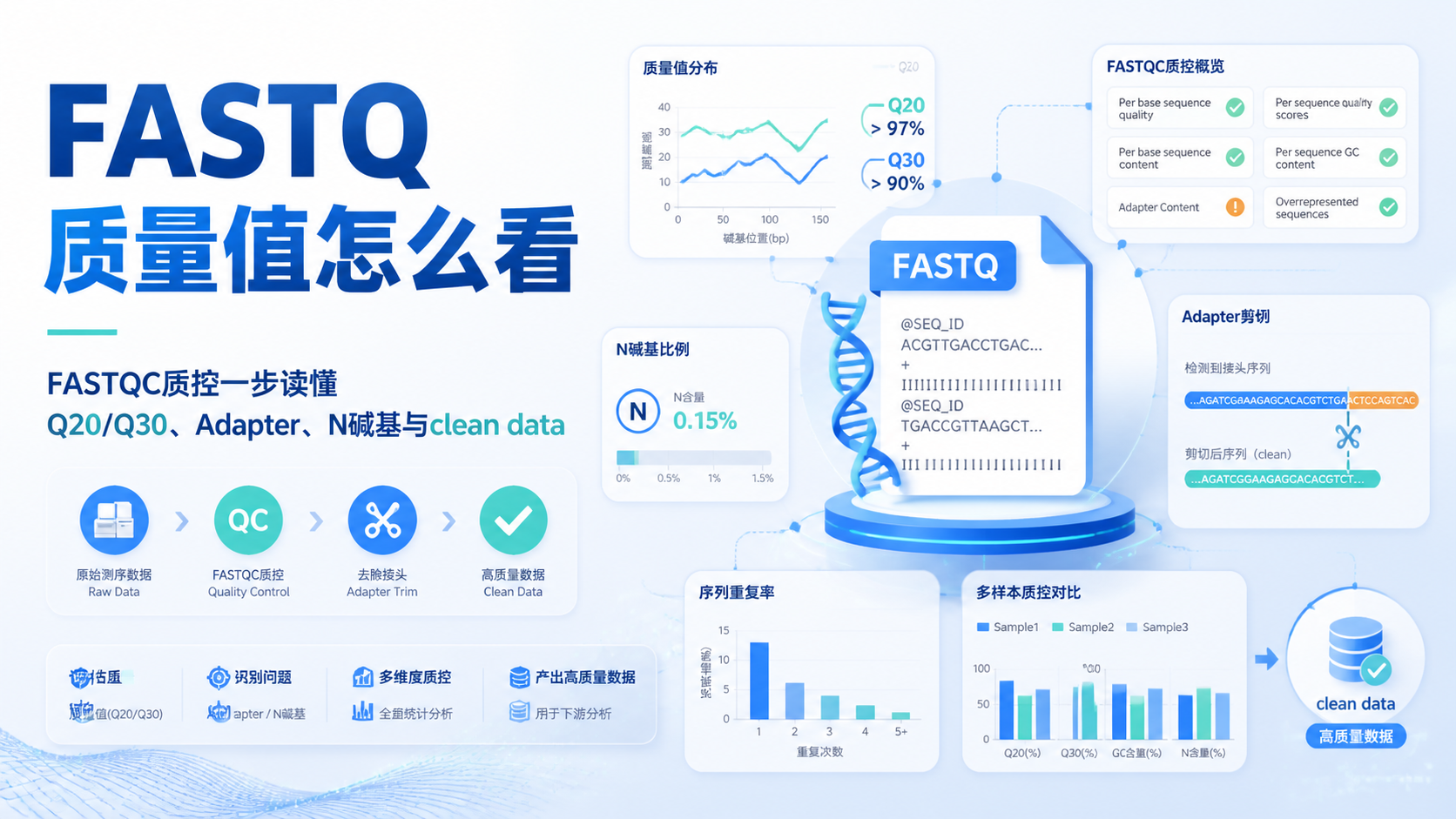
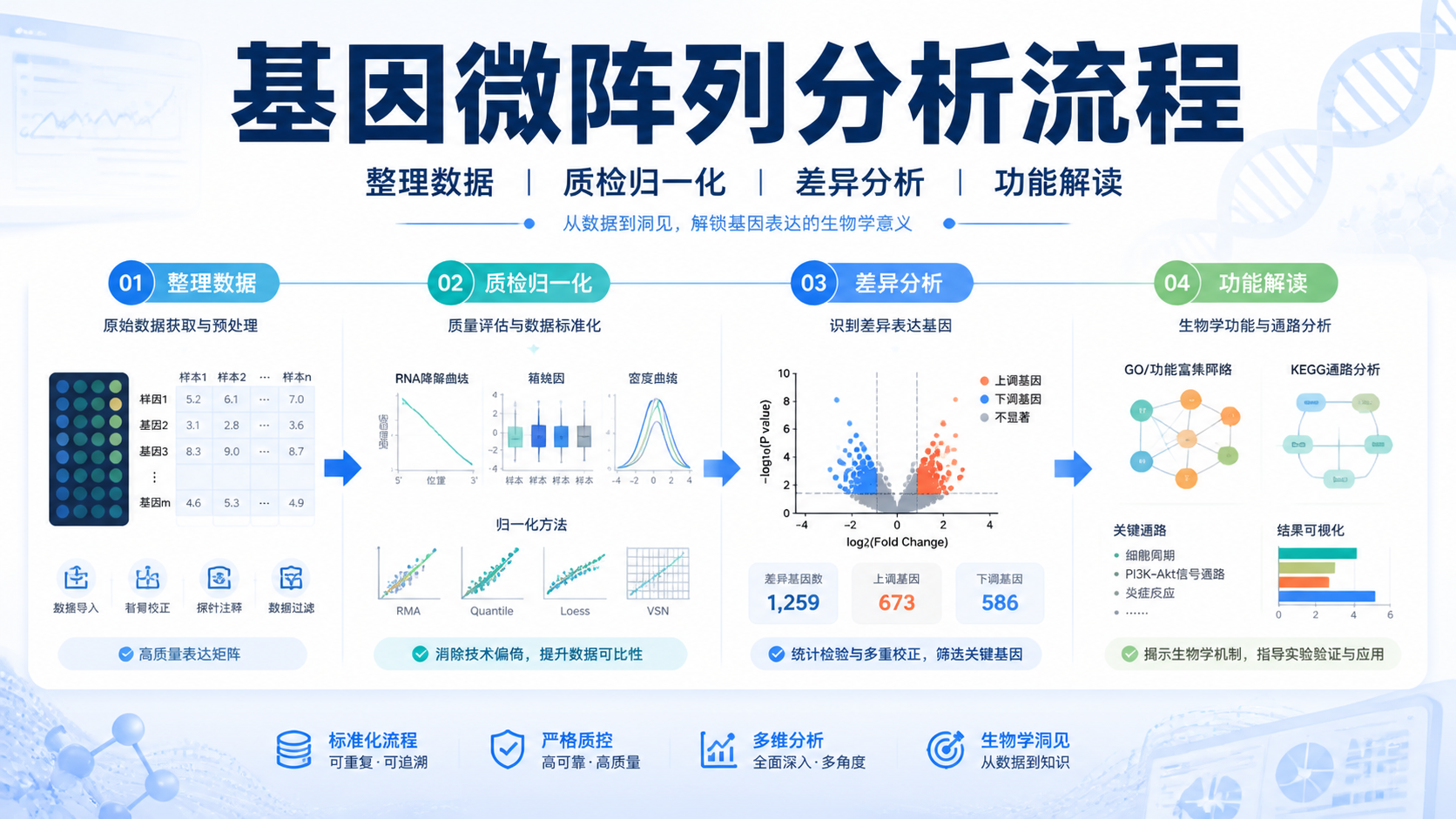
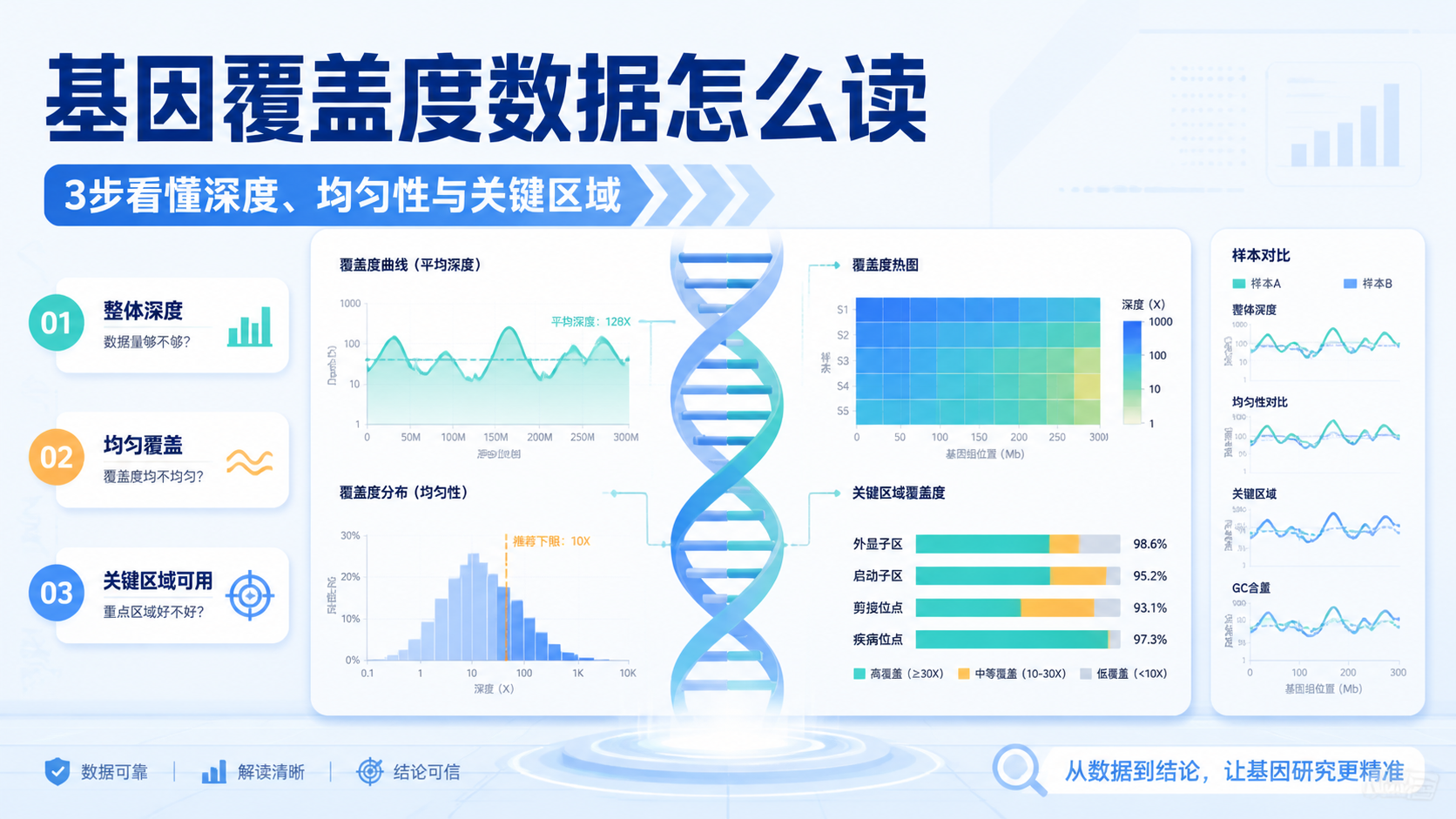
引言Introduction
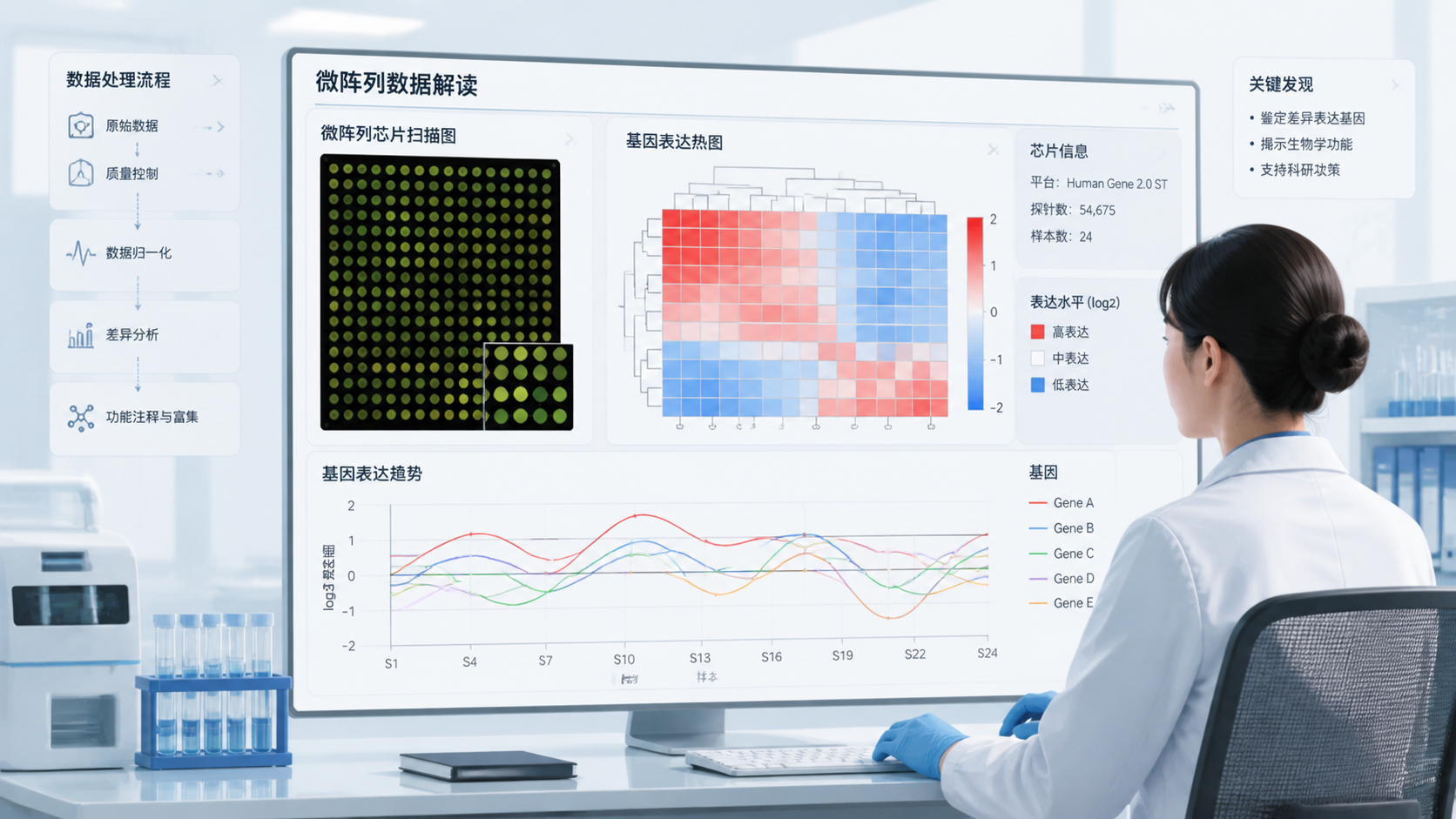
微阵列数据看起来只是几千到几万行的表达值,但真正难点在于,如何从噪声里找到可靠信号,并把结果转化为可复现的结论。对于医学生、医生和科研人员来说,微阵列数据解读 不是单纯看差异倍数,而是一个从质量控制到生物学解释的完整流程。做对了,能提升论文可信度;做错了,常见问题就是假阳性过多、批次效应干扰、结论难以复现。
1. 先把原始微阵列数据“读干净”
1.1 先做质量控制,再谈差异分析
微阵列数据解读的第一步,不是直接找差异基因,而是确认数据是否可信。 原始芯片信号通常受杂交效率、背景噪声、样本质量和扫描参数影响。如果这一步跳过,后面的统计结果再漂亮也可能是假的。
常见的质量控制包括:
- 检查样本整体分布是否一致。
- 观察是否存在离群样本。
- 查看芯片信号强度和背景值。
- 评估探针缺失率和重复性。
在实际研究中,研究者通常会先看箱线图、密度图和PCA图。箱线图能快速看出样本是否整体偏移,PCA图能帮助发现离群点和批次分层。如果同组样本在PCA中明显分散,说明数据稳定性可能不足。
1.2 标准化处理是后续分析的基础
微阵列平台之间、甚至同一批次内,信号强度都可能不完全一致。因此,标准化是微阵列数据解读 中非常关键的一环。它的目标不是“美化数据”,而是让不同样本之间具有可比性。
常见做法包括背景校正、归一化和探针汇总。对于表达谱分析,研究中最常见的是让各样本的整体分布趋于一致,从而减少技术误差对结果的影响。没有标准化的数据,差异分析结果往往会被系统偏差放大。
1.3 批次效应要尽早识别
批次效应是微阵列研究里最容易被忽视的问题之一。它指的是不同实验批次、不同试剂、不同操作人员或不同扫描时间带来的系统性差异。即使生物学分组完全正确,批次效应也可能让样本按“实验时间”而不是按“疾病状态”聚类。
因此,在微阵列数据解读 前期,就应结合实验记录检查:
- 样本是否跨批次混杂。
- 处理组和对照组是否在不同时间完成。
- 是否存在不同平台或不同芯片版本。
- 是否需要在分析前进行批次校正。
这一点对临床样本尤其重要。因为样本量通常不大,一旦批次效应明显,后续统计功效会进一步下降。
2. 用统计方法找出真正有意义的信号
2.1 差异表达分析要关注统计阈值
完成预处理后,才进入微阵列数据解读 的核心环节,也就是差异表达分析。常见做法是比较两组或多组样本的表达差异,并结合P值、FDR和倍数变化筛选候选基因。
在实践中,不能只看倍数变化。因为高表达基因即使变化不大,也可能具有生物学意义;而低表达基因即使倍数很高,也可能不稳定。更稳妥的做法是同时考虑:
- 统计显著性,如P值。
- 多重检验校正,如FDR。
- 生物学效应大小,如log2 fold change。
只有把统计显著性和效应大小放在一起看,微阵列数据解读才更可靠。
2.2 多重比较校正不能省
微阵列一次性检测的探针数量通常很多,常常是上千到上万级别。这样一来,单纯按P<0.05筛选会产生大量假阳性。对于医学研究,这会直接影响结论可信度。
因此,分析时通常要进行多重比较校正,例如控制FDR。这样可以把“看起来显著”但实际不可靠的结果筛掉。如果论文只报告未校正P值,审稿人通常会质疑结果稳定性。
这也是为什么在微阵列数据解读 中,统计方法不是附属步骤,而是决定结论是否成立的关键。
2.3 结果筛选要结合研究场景
不同研究目的,对结果筛选标准也应不同。基础研究可能更重视机制候选基因,临床研究则更关注可转化标志物。若直接套用统一阈值,容易错过真正重要的信号。
建议按以下逻辑筛选:
- 先用统计学阈值获得候选集。
- 再结合表达方向和变化幅度筛选。
- 最后结合文献和疾病背景进行人工判断。
微阵列数据解读的价值,不在于“筛出最多基因”,而在于筛出最值得验证的基因。
3. 把数据结果转成可解释的生物学结论
3.1 功能富集分析帮助建立机制框架
差异基因列表只是起点,不是终点。真正能提升文章质量的,是把这些基因放回通路和功能网络中去理解。常见分析包括GO富集、KEGG通路分析和GSEA等。
这些分析能回答三个问题:
- 这些基因主要参与什么生物过程。
- 它们是否集中在某些信号通路。
- 是否提示特定疾病机制被激活或抑制。
在微阵列数据解读 中,富集分析的意义是把“基因变化”转换为“机制变化”。例如,若炎症相关通路和免疫反应持续富集,就说明研究对象可能存在免疫微环境重塑,而不是单个基因的偶然波动。
3.2 构建网络可以找到关键节点
当候选基因较多时,网络分析往往比单基因罗列更有价值。PPI网络、共表达网络或枢纽基因分析,都能帮助识别可能的关键调控节点。对于科研人员来说,这一步非常重要,因为它有助于从一堆差异基因里提炼出少数优先验证目标。
微阵列数据解读如果只停留在列表层面,结论往往不够深。 通过网络分析,可以进一步筛选出与疾病表型更相关的关键基因、核心通路和潜在药物靶点。
3.3 结果验证决定最终可信度
无论分析结果多漂亮,都需要验证。常见验证方式包括独立队列验证、qPCR验证、文献交叉验证,必要时还可结合蛋白水平或功能实验。对临床研究而言,如果没有验证,微阵列发现通常只能算“候选结果”。
建议至少完成以下验证链条:
- 在独立样本中重复分析。
- 用不同方法验证表达趋势。
- 结合临床表型评估相关性。
- 在讨论中说明局限性。
只有验证闭环完整,微阵列数据解读才具备发表和转化价值。
4. 提高效率的关键:把复杂流程交给专业工具
4.1 常见痛点不是“不会分析”,而是“分析链条太长”
很多医学生和科研人员并不是不会看图,而是卡在流程碎片化。原始数据下载、质控、标准化、差异分析、富集分析、作图和结果整理,往往需要多个工具切换,既耗时,也容易出错。
在微阵列研究中,真正拉开差距的,往往不是某一个算法,而是是否能稳定、规范地完成整条分析链 。这也是为什么不少研究团队会借助专业平台提升效率,减少重复劳动,把更多时间留给实验设计和论文写作。
4.2 解螺旋品牌可以帮你更快完成分析闭环
如果你正在处理微阵列数据 ,又希望结果更清晰、流程更规范,可以考虑使用解螺旋的相关分析与科研支持服务。它的价值不只是“做出图”,而是帮助你把数据从原始矩阵快速推进到可解释、可汇报、可写作的结果阶段。
对研究者而言,这类支持通常能解决几个实际问题:
- 降低预处理和绘图的时间成本。
- 减少标准化和筛选环节的操作失误。
- 更快得到适合论文和汇报的结果图表。
- 让微阵列数据解读 更接近发表要求。
对于时间紧、样本多、又要兼顾论文进度的团队来说,这种专业支持能显著提高效率。
总结Conclusion
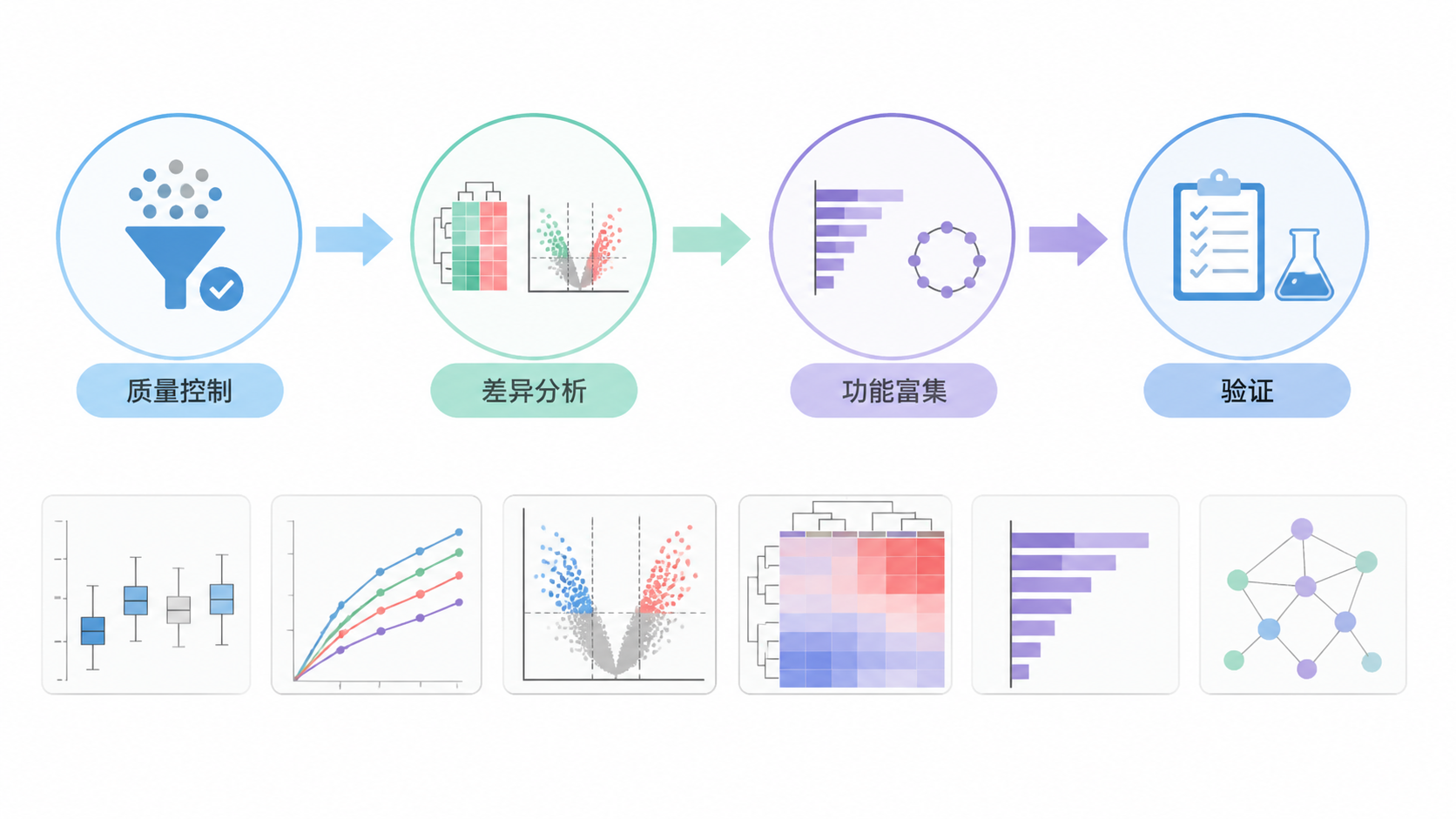
微阵列数据解读的核心,只有三步。 先把数据读干净,再用统计方法找出可靠信号,最后把结果转成可解释的生物学结论。每一步都不能省。因为微阵列研究的价值,不在于数据量大,而在于结论是否可靠、是否可验证、是否能支撑后续研究。
如果你希望更高效地完成微阵列数据 分析,减少重复操作,并让结果更适合论文写作和科研汇报,解螺旋可以提供更专业的支持。让复杂流程变简单,把时间留给更重要的科研判断。
- 引言Introduction
- 1. 先把原始微阵列数据“读干净”
- 2. 用统计方法找出真正有意义的信号
- 3. 把数据结果转成可解释的生物学结论
- 4. 提高效率的关键:把复杂流程交给专业工具
- 总结Conclusion