引言Introduction
FASTA索引是测序数据分析的第一步。很多医学生和科研人员拿到序列文件后,常会卡在“怎么建索引、索引为什么失效、下游分析为何报错”。搞懂FASTA索引,才能让比对、检索和注释更稳定。

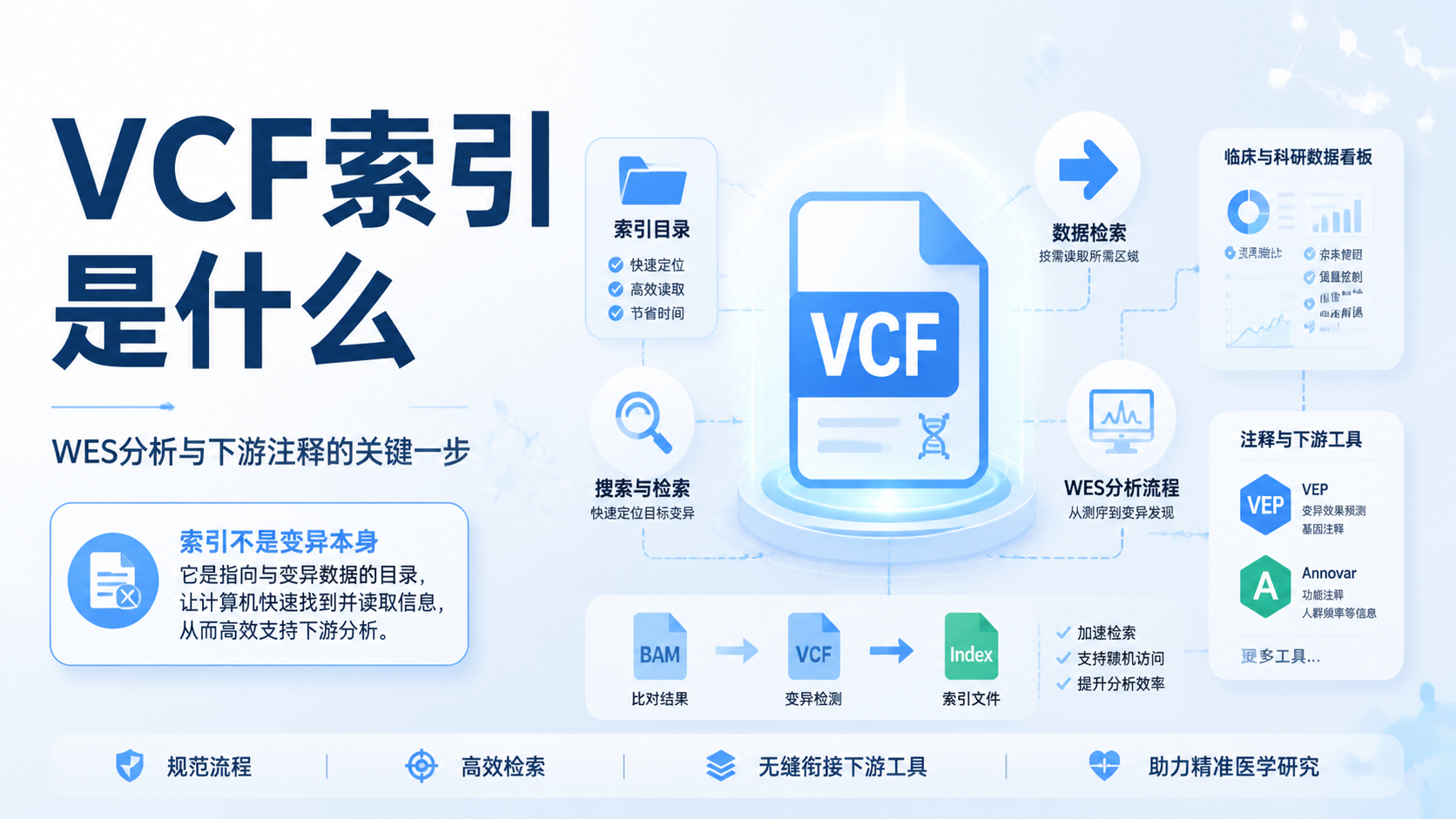
1. 什么是FASTA索引
1.1 FASTA文件与索引的关系
FASTA是生物信息学中最基础的序列格式之一。它以文本形式存储核酸或蛋白序列,常用于数据库检索、比对和下游分析。FASTA索引的核心作用,是让系统快速定位目标序列,而不是逐行扫描整个文件。
从知识库可见,FASTA常作为BLAST组织数据的基本格式,数据库和查询序列都常使用.fa后缀。对于多段序列,还可以合并为multi-FASTA文件,每段序列以前导“>”行区分。这意味着FASTA文件本身是数据容器,索引则是检索入口。
1.2 为什么科研中必须关注索引
在高通量分析中,原始文件通常要先转换为标准序列文件,再进入比对、注释和统计流程。这里最怕两类问题,一类是文件格式不规范,一类是索引与文件不匹配。一旦索引失配,后续分析可能直接失败,或者结果偏差。
高通量检测数据常需要依赖特定仪器和软件流程,后续分析也需要经验。公司可提供基础分析,但个性化需求往往还要自己处理。所以,对FASTA索引的理解,直接影响你能否独立完成数据分析。
2. FASTA索引的4个关键问题
2.1 问题一,FASTA索引到底解决什么
FASTA索引解决的是“快速查找”的问题。序列文件越大,逐条读取越慢。对于含有大量序列的参考库、转录本集合或自建数据库,索引能显著提升访问效率。本质上,它把“遍历搜索”变成“定点定位”。
在测序分析中,这种需求非常常见。比如转录本测序、全基因组重测序、lncRNA测序和small RNA测序,都会产生大量序列信息。如果没有高效索引,下游比对和检索会明显变慢。
2.2 问题二,哪些文件格式最相关
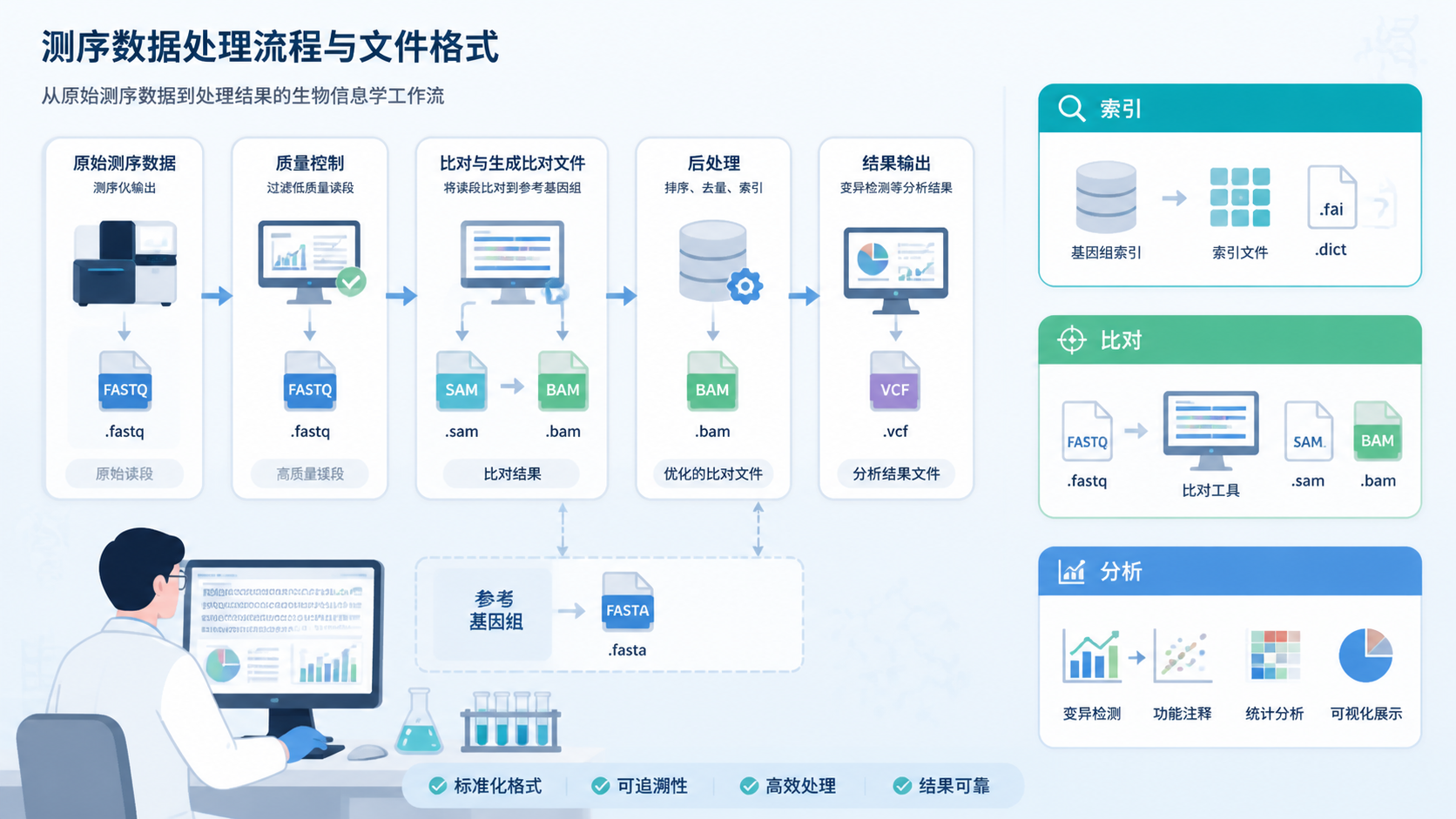
知识库中明确提到,原始序列文件如ab1、SRR、BCL等,通常会先转换成FASTA或FASTQ再处理。FASTQ是最常见的测序原始数据格式,包含4行信息,既有序列正文,也有测序质量。而FASTA更偏向“已整理好的序列表达”。
此外,SAM和BAM也常出现在下游分析中。SAM存储reads到参考序列的比对信息,BAM是其二进制压缩格式,体积更小、检索更快。这说明序列分析中,不同格式承担不同角色,FASTA索引只是其中关键一环。
2.3 问题三,FASTA索引为什么会和下游分析报错有关
下游报错,常见原因不是算法本身,而是输入文件不一致。比如参考FASTA更新了,但索引还是旧版本。或者序列名称、换行、字符编码发生变化。只要FASTA文件内容改变,索引通常也要重新生成。
这类问题在实操中非常隐蔽。表面上文件还能打开,但比对软件读取的是旧索引逻辑。结果就是定位错误、匹配失败,甚至输出空结果。所以,FASTA索引不是一次性操作,而是与文件版本绑定的。
2.4 问题四,如何判断索引是否可用
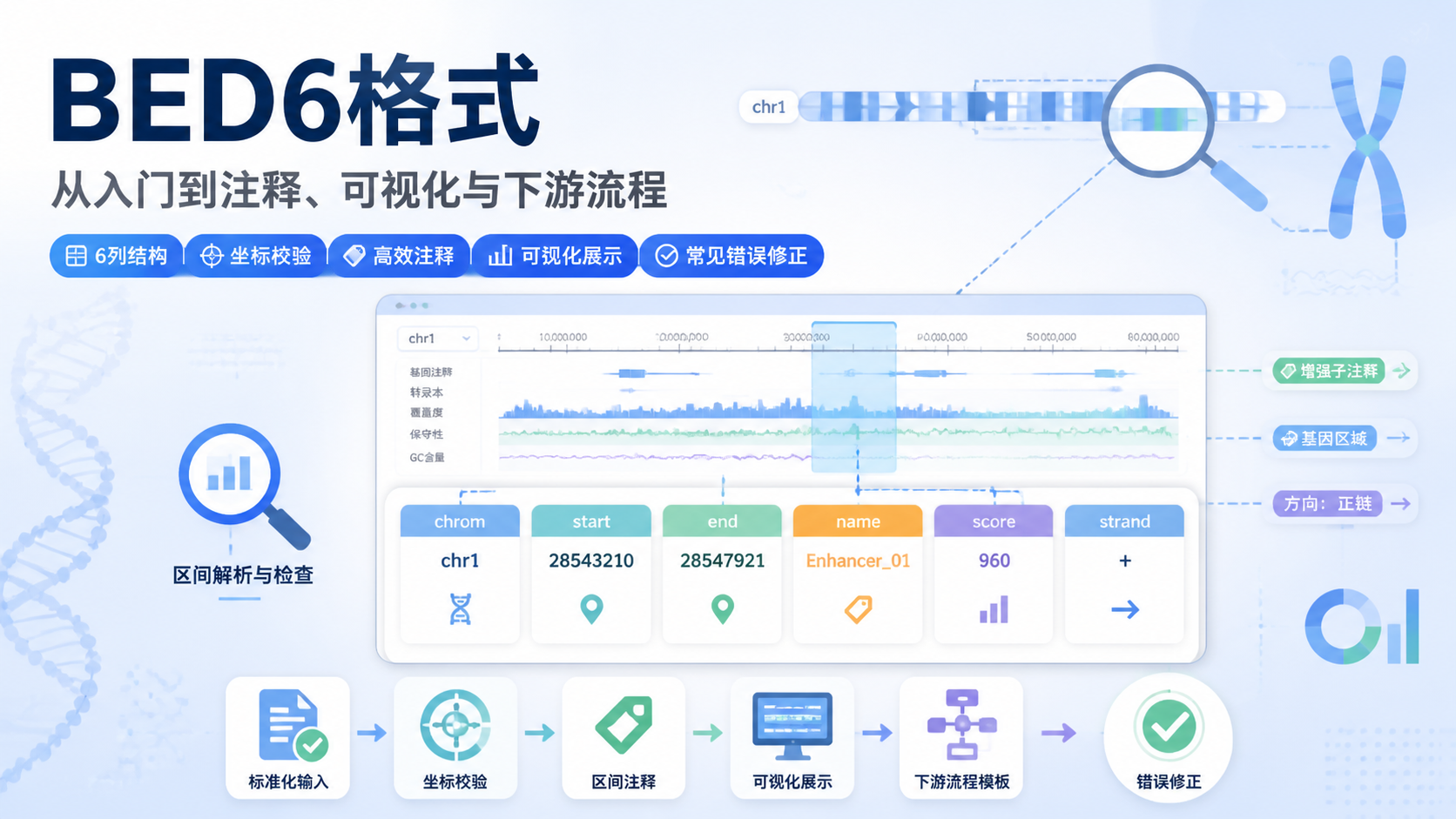
判断FASTA索引是否可用,重点看三件事。
- 文件是否为标准FASTA格式。
- 序列命名是否统一,是否含有特殊字符。
- 索引是否与当前FASTA文件完全对应。
如果文件已修改,哪怕只改了一个序列名,也要重新确认索引。
对科研人员来说,最稳妥的做法是保留原始FASTA、索引文件和分析记录的对应关系,避免版本混乱。
3. FASTA索引在常见高通量场景中的应用
3.1 测序数据分析中的索引需求
高通量测序通过测定核酸序列获得基因变化信息。转录本测序可以研究基因结构、可变剪接和新转录本预测。全基因组重测序可识别SNP、InDel和CNV。这些任务都依赖参考序列的高效读取,因此FASTA索引非常重要。
例如,全基因组重测序本质上要把不同个体的基因组序列与参考序列进行比对。参考序列越大,索引价值越高。没有索引,分析时间和资源消耗都会明显增加。
3.2 与芯片、测序和序列文件的衔接
知识库中提到,目前较成熟且应用广的高通量技术主要有芯片和测序。芯片侧重核酸或蛋白水平的检测,测序则直接读取序列信息。当数据进入测序分支后,FASTA索引就是基础设施。
FASTA/FASTQ/SAM/BAM这些格式之间存在清晰分工。FASTA便于组织序列,FASTQ保留原始测序质量,SAM记录比对结果,BAM则提升存储和检索效率。理解这条链路,才能真正理解FASTA索引在流程中的位置。
3.3 转录因子和数据库检索中的扩展价值
在转录因子研究中,JASPAR数据库可用于检索转录因子结合位点,并支持预测调控基因的转录因子。这类数据库工作虽然不直接等同于FASTA索引,但其底层逻辑同样依赖标准化序列管理。
例如,数据库检索中常需要明确种属、位点版本和序列信息。序列数据越规范,后续比对和预测越可靠。 这也是为什么从基础文件开始做好索引和命名,能减少很多后续问题。
4. 实操建议:如何减少FASTA索引相关问题
4.1 建库前先统一文件规范
在生成FASTA索引前,先检查文件头、序列名和内容格式。避免同一项目中出现多个命名体系。建议统一使用简洁、稳定、无特殊符号的序列ID。
如果你的项目包含多个样本或多个参考版本,最好建立清晰的目录结构。原始文件、处理后文件、索引文件分别存放。这样可以降低版本混用风险。
4.2 文件更新后立即重建索引
只要FASTA内容改动,就要重新确认索引是否需要更新。包括新增序列、删除序列、修改序列名、替换参考版本。不要默认旧索引还能继续用。
这是最常见也最容易被忽略的错误。很多分析失败,不是因为参数错,而是因为输入文件和索引文件不是同一版本。这一点对临床科研和组学分析都很关键。
4.3 结合下游工具理解索引意义
从知识库看,测序分析常会接触FASTA、FASTQ、SAM和BAM。不同文件有不同用途。学会区分“原始数据”“序列数据”“比对数据”,就能更准确地判断索引应放在哪一步。
如果你在进行BLAST检索、参考基因组比对或转录本分析,优先检查参考FASTA是否规范。这能显著减少重复排错时间。
总结Conclusion
FASTA索引看似只是一个基础步骤,实际却关系到序列检索效率、文件一致性和下游分析稳定性。本文围绕4个关键问题,解释了FASTA索引的作用、相关文件格式、常见报错原因和实操建议。对医学生、医生和科研人员来说,先把基础打牢,才能让高通量分析走得更稳。
如果你希望进一步提升文献检索、数据库使用和科研数据分析效率,可以借助解螺旋 的科研技能与实操课程,系统补齐从序列文件到下游分析的关键能力。把复杂流程拆成标准步骤,才能更快解决实际问题。
- 引言Introduction
- 1. 什么是FASTA索引
- 2. FASTA索引的4个关键问题
- 3. FASTA索引在常见高通量场景中的应用
- 4. 实操建议:如何减少FASTA索引相关问题
- 总结Conclusion