





资讯
 CNV数据正在成为肿瘤和遗传研究中提升结论可信度的重要证据。很多课题只做转录组,容易停留在“表达变化”。如果加入CNV数据,就能从拷贝数层面解释基因异常,减少单一组学带来的偏差。阅读25 · 点赞0Dr.Sheng 于2026-05-12发布
CNV数据正在成为肿瘤和遗传研究中提升结论可信度的重要证据。很多课题只做转录组,容易停留在“表达变化”。如果加入CNV数据,就能从拷贝数层面解释基因异常,减少单一组学带来的偏差。阅读25 · 点赞0Dr.Sheng 于2026-05-12发布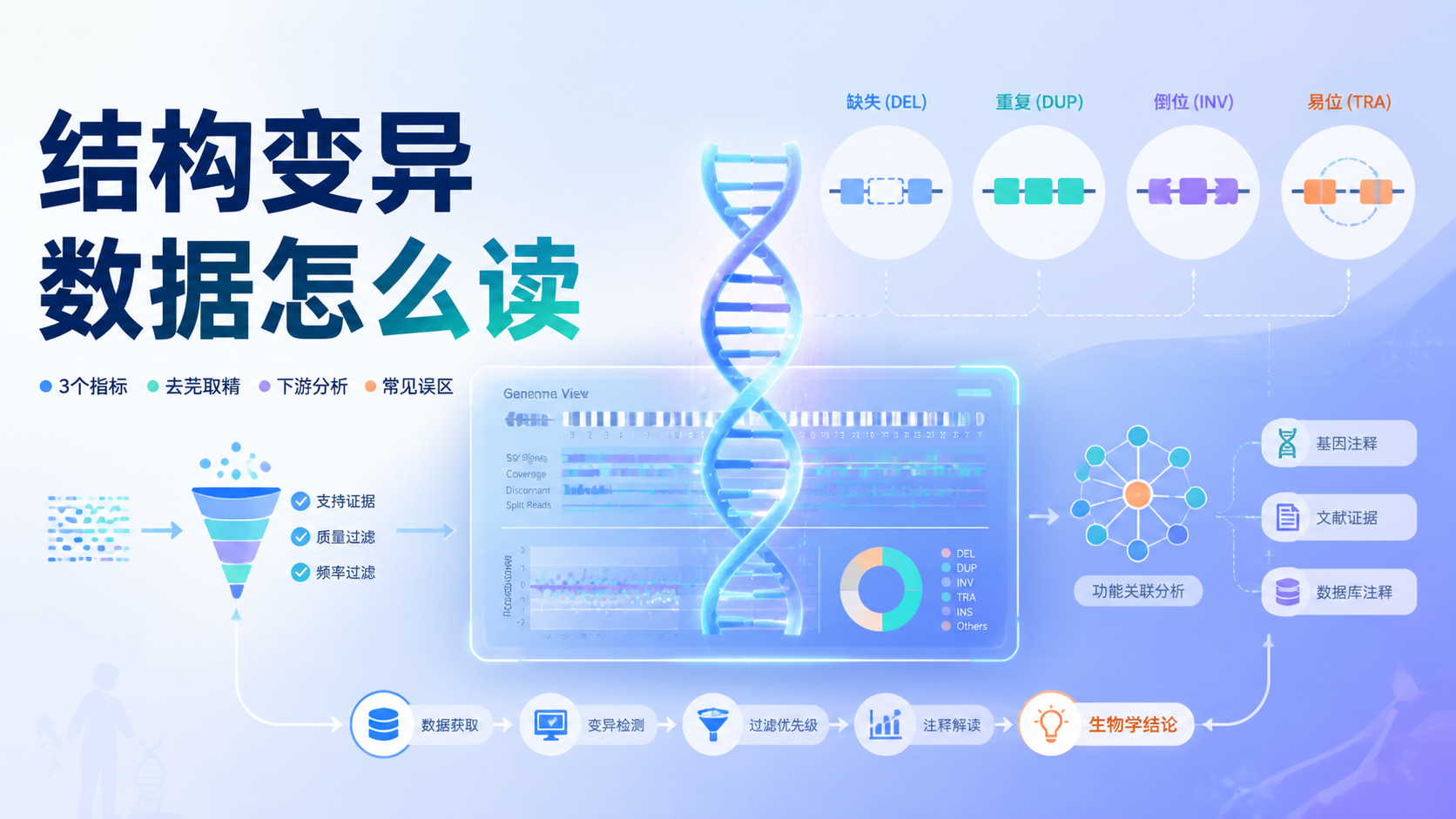 在科研里,结构变异数据常让人“看见了信号,却抓不住重点”。数据量大、差异多、下游解释难,是很多医学生、医生和科研人员的共同痛点。本文用5个关键点,帮你把结构变异数据从“结果表”读成“研究线索”。阅读6 · 点赞0Dr.Sheng 于2026-05-12发布
在科研里,结构变异数据常让人“看见了信号,却抓不住重点”。数据量大、差异多、下游解释难,是很多医学生、医生和科研人员的共同痛点。本文用5个关键点,帮你把结构变异数据从“结果表”读成“研究线索”。阅读6 · 点赞0Dr.Sheng 于2026-05-12发布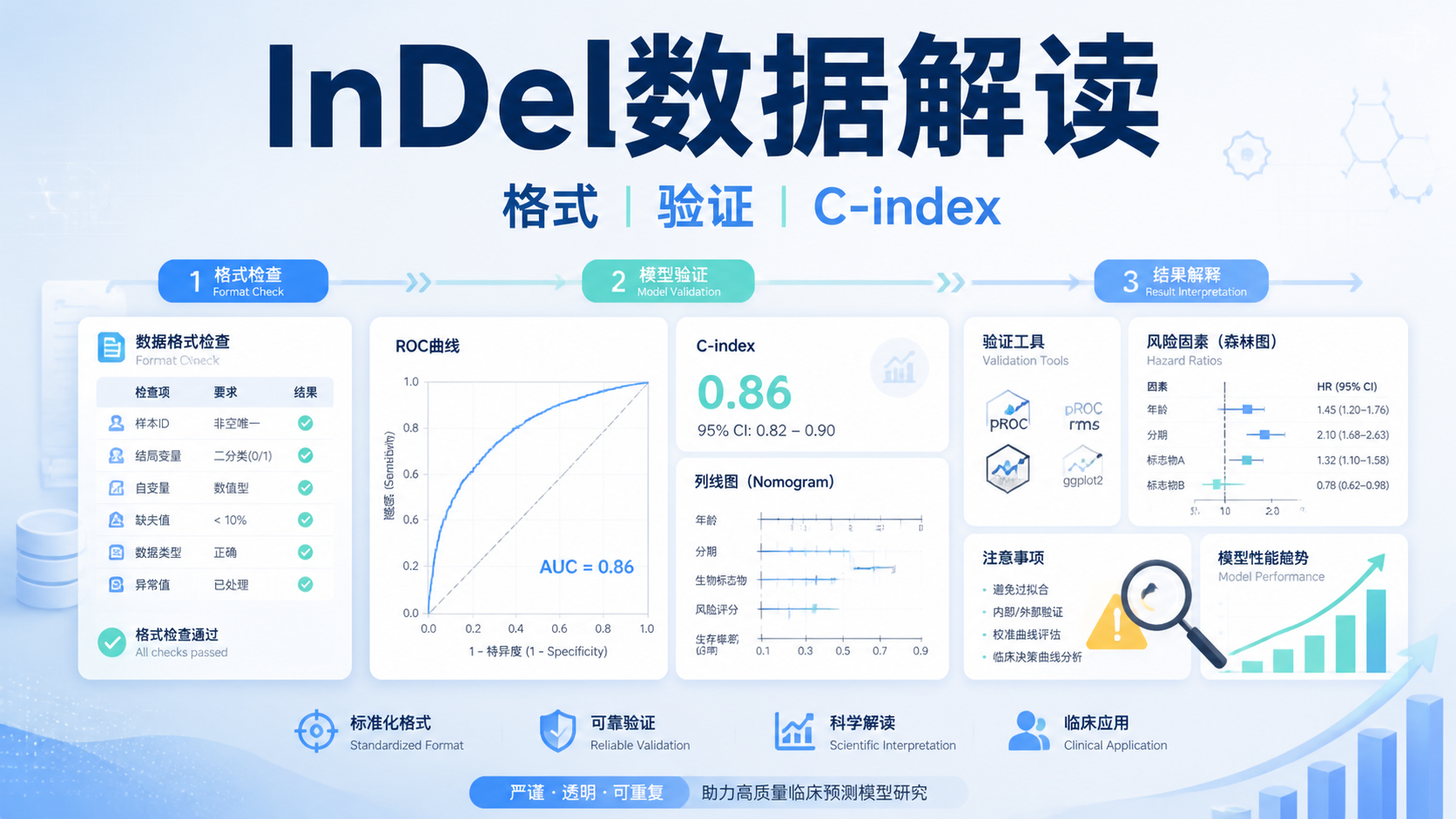 InDel数据解读常见的问题,是样本上传后格式不对、结果偏差大,甚至无法通过验证。对医学生、医生和科研人员来说,真正影响结论的,往往不是算法本身,而是数据规范、字段一致性和结果验证是否到位。阅读4 · 点赞0Dr.Sheng 于2026-05-12发布
InDel数据解读常见的问题,是样本上传后格式不对、结果偏差大,甚至无法通过验证。对医学生、医生和科研人员来说,真正影响结论的,往往不是算法本身,而是数据规范、字段一致性和结果验证是否到位。阅读4 · 点赞0Dr.Sheng 于2026-05-12发布 SNP数据分析看似流程固定,真正难点却在选题、工具变量、数据协调、稳健性和工具软件。任何一步出错,后面的因果推断都会失真。本文按孟德尔随机化的研究框架,拆解SNP数据分析的5个核心问题,帮助医学生、医生和科研人员快速建立正确思路。阅读4 · 点赞0Dr.Sheng 于2026-05-12发布
SNP数据分析看似流程固定,真正难点却在选题、工具变量、数据协调、稳健性和工具软件。任何一步出错,后面的因果推断都会失真。本文按孟德尔随机化的研究框架,拆解SNP数据分析的5个核心问题,帮助医学生、医生和科研人员快速建立正确思路。阅读4 · 点赞0Dr.Sheng 于2026-05-12发布
 转录本注释文件是做转录组分析、差异表达和机制研究时绕不开的基础文件。很多人明明有测序数据,却因为注释版本不一致、坐标不匹配、基因与转录本关系混乱,导致结果反复重跑。阅读4 · 点赞0Dr.Sheng 于2026-05-12发布
转录本注释文件是做转录组分析、差异表达和机制研究时绕不开的基础文件。很多人明明有测序数据,却因为注释版本不一致、坐标不匹配、基因与转录本关系混乱,导致结果反复重跑。阅读4 · 点赞0Dr.Sheng 于2026-05-12发布 基因注释文件看似只是背景资料,实则直接影响变异定位、功能解释和下游统计。对医学生、医生和科研人员来说,同一个变异,因注释文件不同,结论可能完全不同。这也是精准分析中最容易被忽视、却最容易出错的一步。阅读4 · 点赞0Dr.Sheng 于2026-05-12发布
基因注释文件看似只是背景资料,实则直接影响变异定位、功能解释和下游统计。对医学生、医生和科研人员来说,同一个变异,因注释文件不同,结论可能完全不同。这也是精准分析中最容易被忽视、却最容易出错的一步。阅读4 · 点赞0Dr.Sheng 于2026-05-12发布 在科研写作和投稿流程中,FQ格式常被用来承载关键信息,但很多人不是写不全,就是格式不统一,导致检索、审核和沟通效率下降。掌握FQ格式,核心不是“写得多”,而是“写得准、写得规范”。阅读10 · 点赞0Dr.Sheng 于2026-05-12发布
在科研写作和投稿流程中,FQ格式常被用来承载关键信息,但很多人不是写不全,就是格式不统一,导致检索、审核和沟通效率下降。掌握FQ格式,核心不是“写得多”,而是“写得准、写得规范”。阅读10 · 点赞0Dr.Sheng 于2026-05-12发布 在论文投稿、课题申报和实验报告中,FA格式 常被写错。格式不统一,会直接影响审稿效率、文档专业度,甚至让引用信息失真。对于医学生、医生和科研人员来说,掌握FA格式 不是细节问题,而是基础能力。阅读7 · 点赞0Dr.Sheng 于2026-05-12发布
在论文投稿、课题申报和实验报告中,FA格式 常被写错。格式不统一,会直接影响审稿效率、文档专业度,甚至让引用信息失真。对于医学生、医生和科研人员来说,掌握FA格式 不是细节问题,而是基础能力。阅读7 · 点赞0Dr.Sheng 于2026-05-12发布 GTF2格式是生信注释里最常见的文件之一,但很多人第一次接触时,都会卡在“怎么读、怎么用、怎么和表达矩阵对应”这一步。如果你也在做RNA-seq、基因注释或下游差异分析,先读懂GTF2格式,能少走很多弯路。阅读1 · 点赞0Dr.Sheng 于2026-05-12发布
GTF2格式是生信注释里最常见的文件之一,但很多人第一次接触时,都会卡在“怎么读、怎么用、怎么和表达矩阵对应”这一步。如果你也在做RNA-seq、基因注释或下游差异分析,先读懂GTF2格式,能少走很多弯路。阅读1 · 点赞0Dr.Sheng 于2026-05-12发布 GFF3格式是基因组注释中最常见的数据组织方式之一。很多医学生、医生和科研人员在处理测序结果时,都会遇到“看得见数据,却读不懂注释”的问题。如果不理解GFF3格式,就很难高效完成基因定位、功能注释和下游分析。阅读1 · 点赞0Dr.Sheng 于2026-05-12发布
GFF3格式是基因组注释中最常见的数据组织方式之一。很多医学生、医生和科研人员在处理测序结果时,都会遇到“看得见数据,却读不懂注释”的问题。如果不理解GFF3格式,就很难高效完成基因定位、功能注释和下游分析。阅读1 · 点赞0Dr.Sheng 于2026-05-12发布
 SAM格式是生物医学和组学分析里最常见的比对结果格式之一。很多医学生、医生和科研人员在看测序结果时,常会卡在“字段太多、含义不清、不会判断质量”这一步。掌握SAM格式怎么写,不只是会排版,更是理解比对结果的基础。阅读1 · 点赞0Dr.Sheng 于2026-05-12发布
SAM格式是生物医学和组学分析里最常见的比对结果格式之一。很多医学生、医生和科研人员在看测序结果时,常会卡在“字段太多、含义不清、不会判断质量”这一步。掌握SAM格式怎么写,不只是会排版,更是理解比对结果的基础。阅读1 · 点赞0Dr.Sheng 于2026-05-12发布 GTF文件注释是转录组、外显子组和变异分析的基础。很多人做完测序后,卡在“结果能不能信”这一步。问题往往不在测序本身,而在注释文件是否选对、是否用对。一份GTF文件注释,决定了基因、转录本和突变位置能否被准确解释。阅读2 · 点赞0Dr.Sheng 于2026-05-12发布
GTF文件注释是转录组、外显子组和变异分析的基础。很多人做完测序后,卡在“结果能不能信”这一步。问题往往不在测序本身,而在注释文件是否选对、是否用对。一份GTF文件注释,决定了基因、转录本和突变位置能否被准确解释。阅读2 · 点赞0Dr.Sheng 于2026-05-12发布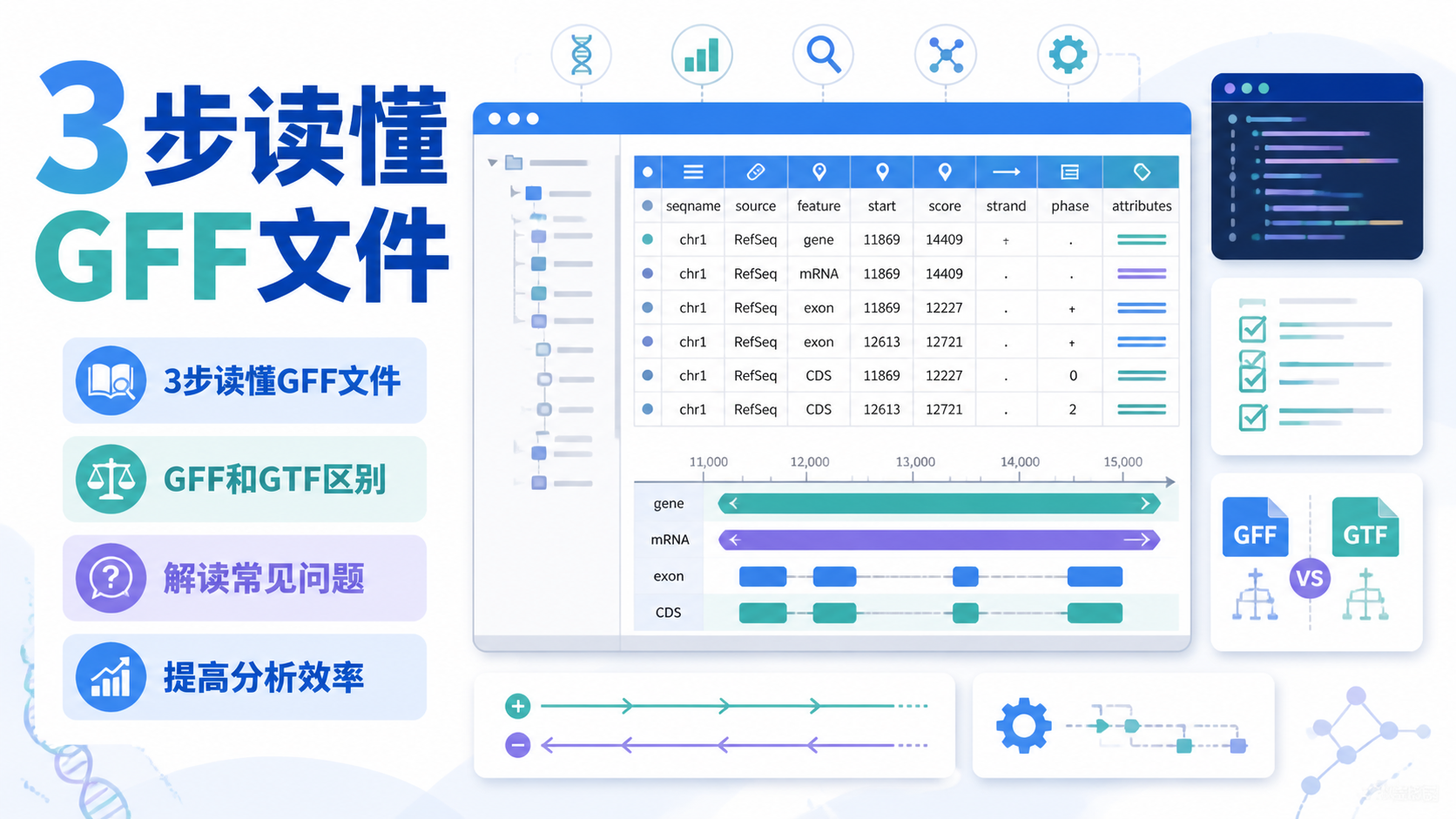 GFF文件解读对很多医学生、医生和科研人员来说并不陌生,但真正上手时,常卡在“字段太多、层级太乱、信息看不懂”。如果你也在做基因注释、转录本分析或可视化,GFF文件解读就是必须掌握的基础技能。阅读2 · 点赞0Dr.Sheng 于2026-05-12发布
GFF文件解读对很多医学生、医生和科研人员来说并不陌生,但真正上手时,常卡在“字段太多、层级太乱、信息看不懂”。如果你也在做基因注释、转录本分析或可视化,GFF文件解读就是必须掌握的基础技能。阅读2 · 点赞0Dr.Sheng 于2026-05-12发布
 BAM文件处理是RNA-seq下游分析的基础。很多人已经拿到比对结果,却卡在格式转换、排序、索引和定量准备上。如果BAM文件没有处理规范,后续表达量分析和可视化都会出错。阅读2 · 点赞0Dr.Sheng 于2026-05-12发布
BAM文件处理是RNA-seq下游分析的基础。很多人已经拿到比对结果,却卡在格式转换、排序、索引和定量准备上。如果BAM文件没有处理规范,后续表达量分析和可视化都会出错。阅读2 · 点赞0Dr.Sheng 于2026-05-12发布 FASTA文件解析是生物信息学分析的第一步,但很多医学生和科研人员常卡在格式细节、批量处理和后续比对衔接上。如果解析不严谨,后面的BLAST、比对和注释都会出错。阅读10 · 点赞0Dr.Sheng 于2026-05-12发布
FASTA文件解析是生物信息学分析的第一步,但很多医学生和科研人员常卡在格式细节、批量处理和后续比对衔接上。如果解析不严谨,后面的BLAST、比对和注释都会出错。阅读10 · 点赞0Dr.Sheng 于2026-05-12发布 患者临床数据不是简单的病历记录。对医学生、医生和科研人员来说,它决定了诊疗总结、科研质量和证据转化效率。如果数据零散、缺失或难以追溯,后续分析和发文都会受影响。阅读7 · 点赞0Dr.Xin 于2026-05-12发布
患者临床数据不是简单的病历记录。对医学生、医生和科研人员来说,它决定了诊疗总结、科研质量和证据转化效率。如果数据零散、缺失或难以追溯,后续分析和发文都会受影响。阅读7 · 点赞0Dr.Xin 于2026-05-12发布
热门文章
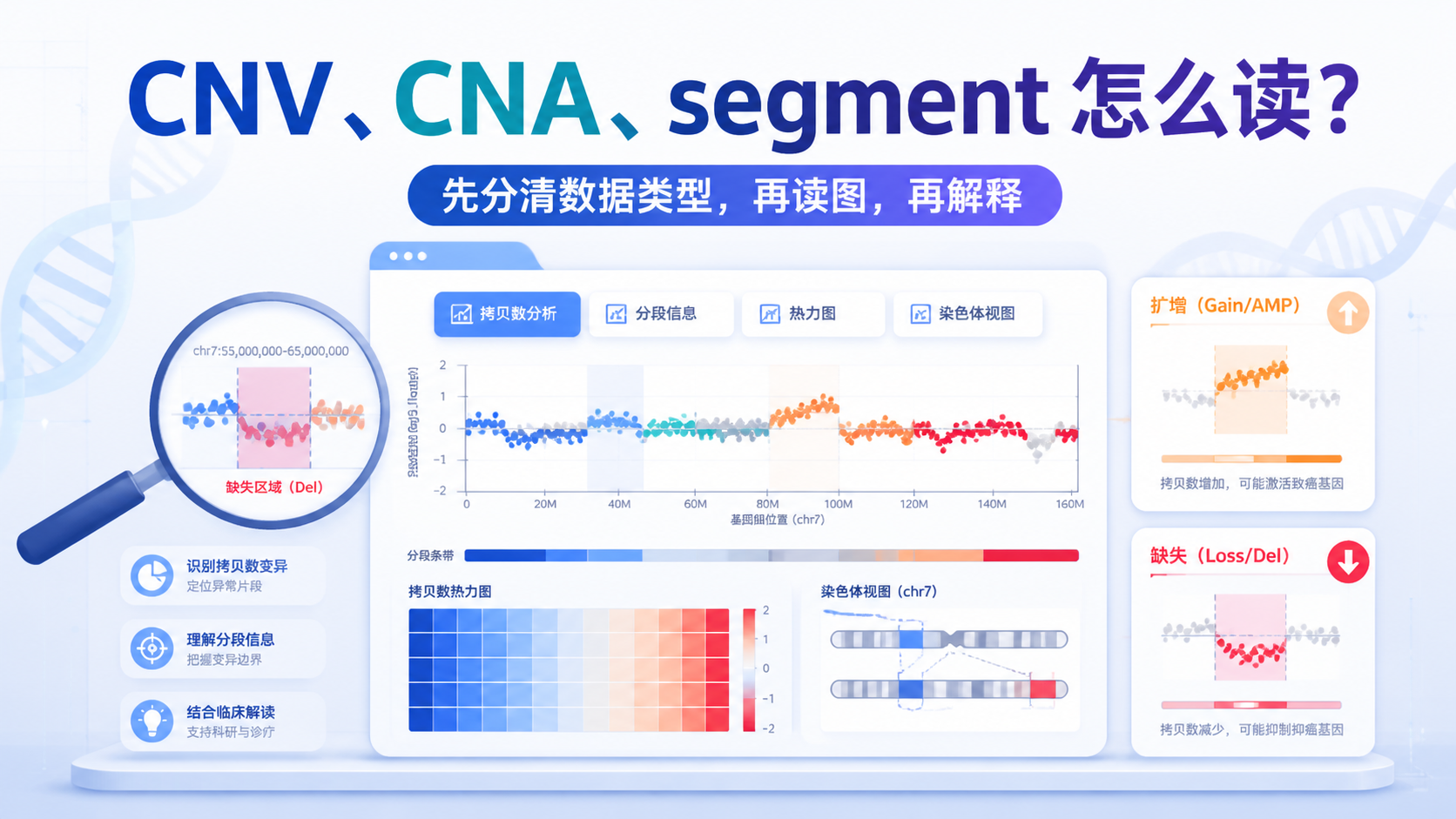
拷贝数变异数据如何解读?3步讲清
阅读40 · 点赞0
生信遗传学基础怎么学?6个高效方法
阅读29 · 点赞0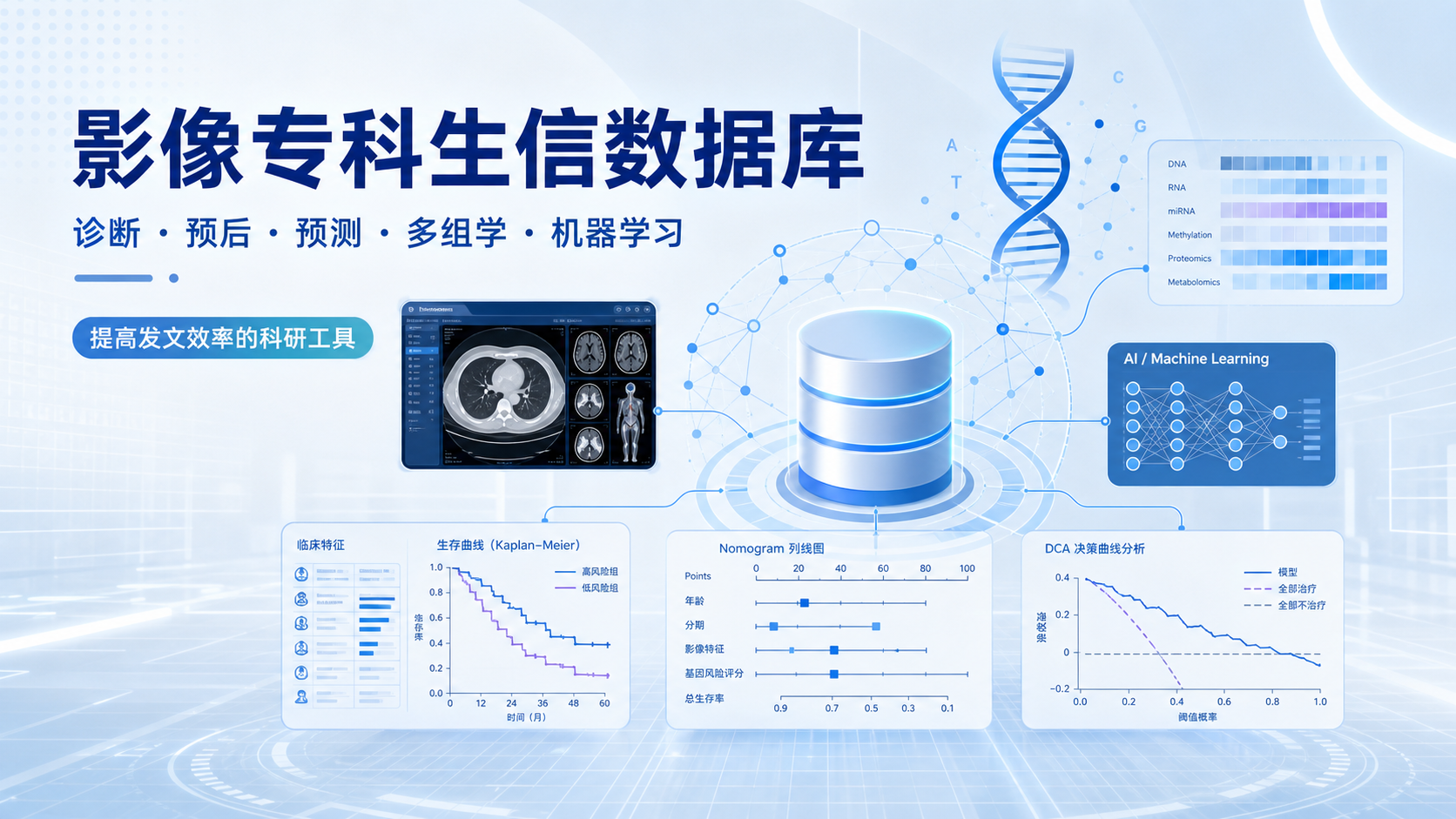
影像专科生信数据库:7个关键应用
阅读26 · 点赞0