引言Introduction
BAM文件处理是RNA-seq下游分析的基础。很多人已经拿到比对结果,却卡在格式转换、排序、索引和定量准备上。如果BAM文件没有处理规范,后续表达量分析和可视化都会出错。

1.BAM文件处理的第一步:理解SAM与BAM的关系
1.1 SAM是原始比对结果
在RNA-seq流程里,Hisat2等比对软件完成后,通常先生成SAM文件。SAM是Sequence Alignment/Map文件,主要用于存放比对信息。它记录每条read与参考基因组的对应关系。
BAM文件处理的起点,不是直接分析BAM,而是先确认SAM是否生成正确。 如果比对率较高,例如课程中提到的96.47%,通常说明数据质量较好,进入下一步的价值更高。
1.2 BAM是SAM的二进制形式
BAM是SAM的二进制压缩格式。它更小,读取更快,也更适合后续工具调用。对于大规模测序数据,这是标准中间文件格式。
从实际工作看,BAM文件处理的核心目标是把可读但臃肿的SAM,转换成更高效、更适合计算的BAM。 这一步是后续排序、索引、计数分析的前提。
2.BAM文件处理的第二步:用Samtools完成格式转换
2.1 samtools view是最常用命令
课程中使用的是Samtools。它是处理比对文件的核心工具,常用功能包括view、sort和index。
在格式转换阶段,主要使用samtools view。
典型思路是:
-S表示输入为SAM格式-b表示输出为BAM格式
例如把56.sam转成56.bam,本质上就是完成一次标准化的格式转换。
BAM文件处理最容易忽略的点,是要先确认文件路径和输入类型是否正确。 路径错了,后面所有步骤都会失败。
2.2 后台运行更适合大文件
RNA-seq数据量通常很大。转换时可以用nohup把任务放到后台运行。这样即使关闭终端,任务也能继续执行。再用jobs查看运行状态,确认进度。
这种做法在真实分析中非常实用。因为一个样本往往不止几百MB,甚至是几十GB。BAM文件处理必须兼顾效率和稳定性。
3.BAM文件处理的第三步:按染色体位置排序
3.1 sort是BAM分析的关键动作
生成BAM后,下一步通常是排序。Samtools的sort会按照染色体位置对比对结果重新排列。排序后的文件一般命名为sorted.bam。
这是非常关键的一步。因为很多下游工具要求BAM文件必须有序。没有排序的文件,后续定量、变异检测、可视化都可能无法运行。
BAM文件处理里,排序不是可选项,而是标准步骤。
3.2 为什么排序这么重要
排序后,数据访问更快,索引也更容易建立。对于基因组浏览器和定量软件来说,排序后的BAM更适合随机读取某个染色体区段。
课程中提到,排序依据是染色体位置。这与常见的转录组分析流程一致。如果你在BAM文件处理时跳过排序,往往会在下游环节补回错误成本。
4.BAM文件处理的第四步:建立索引并管理文件
4.1 index让BAM可快速定位
BAM文件排序后,通常要建立索引。Samtools的index会生成索引文件,便于快速定位到某个基因区域。
这一步对可视化尤其重要。比如在IGV中查看某个区域的比对情况,没有索引就很难高效加载。BAM文件处理做到索引,才算真正具备下游分析条件。
4.2 文件管理同样重要
课程中还提到,将生成的BAM文件从align_tool移动或复制到align目录。这个细节很重要。分析目录混乱,会影响批量处理、复核和结果追踪。
建议按样本建立统一命名规则:
- 原始SAM文件
- 转换后的BAM文件
- 排序后的sorted.bam
- 索引文件.bai
规范命名是BAM文件处理里最便宜、也最有效的质量控制。
5.BAM文件处理的第五步:为定量分析做好准备
5.1 定量前要确认参考基因组和注释文件
课程明确指出,BAM文件处理完成后,下一步就是定量。常见定量软件包括HTSeq count、featureCounts、subread等。
但定量不只看BAM。还必须有:
- 参考基因组
- 注释文件,常见为GTF或GFF
参考基因组提供序列背景,注释文件告诉软件每段比对落在哪个基因或外显子上。没有正确的注释信息,BAM文件处理再规范,定量结果也没有意义。
5.2 从BAM到表达值的逻辑
排序后的BAM会进入read计数流程。常见分析层面包括:
- 基因水平
- 转录本水平
- 外显子水平
其中,基因水平定量最常用。 因为它最适合常规差异表达分析,也最容易被项目和公司报告采用。
从实际分析角度看,BAM文件处理完成后,你得到的不是终点,而是一个可计算、可解释、可复现的输入文件。
6.BAM文件处理中的常见错误与检查要点
6.1 常见错误
实际工作中,BAM文件处理常见问题包括:
- SAM与BAM格式参数写反
- 路径错误,文件找不到
- 未排序就进入定量
- 缺少索引文件
- 命名混乱,样本对应不清
这些问题看似基础,但非常常见。尤其在批量样本分析中,一个错误可能会放大成整批结果偏差。
6.2 建议的检查顺序
建议按以下顺序检查:
- 比对结果是否生成SAM
- SAM是否成功转成BAM
- BAM是否已排序
- 是否已建立索引
- 是否匹配参考基因组与注释文件
把BAM文件处理做成标准流程,比临时修错更重要。
7.如何借助解螺旋提升BAM文件处理效率
7.1 适合科研和教学场景的工具化流程
对于医学生、医生和科研人员来说,真正的痛点往往不是“知道概念”,而是“能不能快速把流程跑通”。BAM文件处理涉及命令行、目录管理、后台任务和下游定量,任何一步出错都会拖慢项目进度。
这也是很多人需要标准化工具和教程支持的原因。解螺旋品牌的价值,就在于帮助用户把复杂流程拆成可执行步骤。
7.2 用标准化流程减少返工
如果你希望减少SAM转BAM、排序、索引和定量准备中的重复试错,可以优先采用成熟、清晰的流程模板。这样能明显降低操作成本,提高数据分析效率。
在真实项目中,规范化的BAM文件处理流程,往往比“临时补救”更可靠。 这也是科研结果可重复、可追踪、可交付的基础。
总结Conclusion
BAM文件处理看似只是格式转换,实际上是RNA-seq分析链条中的关键中间环节。核心步骤可以概括为五点:理解SAM与BAM关系、用Samtools完成转换、按染色体位置排序、建立索引并规范管理文件、为定量分析准备参考基因组和注释信息。
只要这五步做规范,后续表达量分析的稳定性会明显提升。
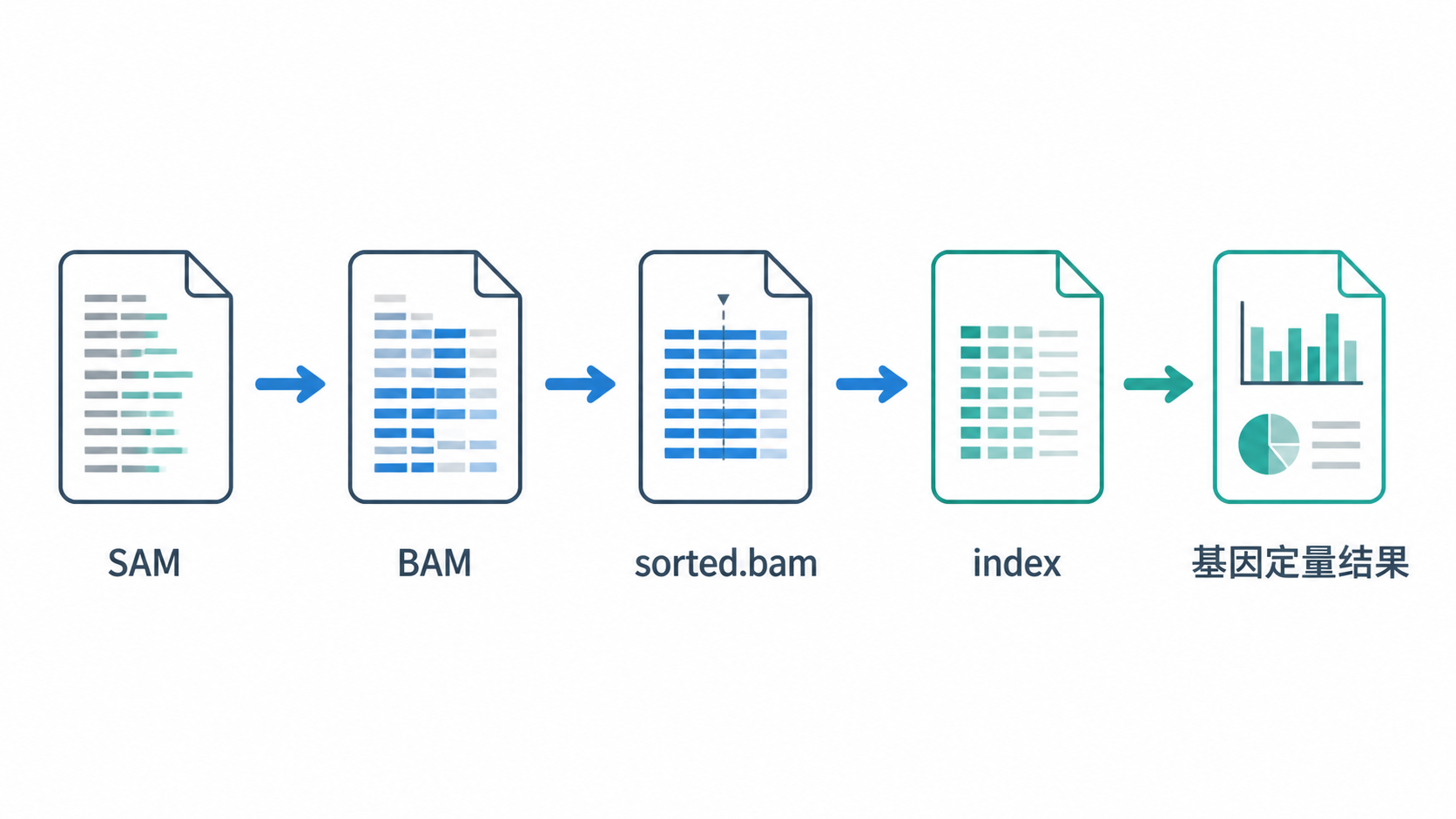
如果你希望把BAM文件处理流程做得更高效、更标准,欢迎关注解螺旋。借助解螺旋的课程与工具支持,你可以更快完成从比对文件到定量结果的关键转换。
- 引言Introduction
- 1.BAM文件处理的第一步:理解SAM与BAM的关系
- 2.BAM文件处理的第二步:用Samtools完成格式转换
- 3.BAM文件处理的第三步:按染色体位置排序
- 4.BAM文件处理的第四步:建立索引并管理文件
- 5.BAM文件处理的第五步:为定量分析做好准备
- 6.BAM文件处理中的常见错误与检查要点
- 7.如何借助解螺旋提升BAM文件处理效率
- 总结Conclusion