引言Introduction
VCF文件注释是WES和肿瘤基因检测里最关键的一步。没有注释,变异只是坐标和碱基替换,无法直接对应基因功能、疾病风险和用药证据。对医学生、医生和科研人员来说,真正的难点不在“有没有变异”,而在“这个变异意味着什么”。
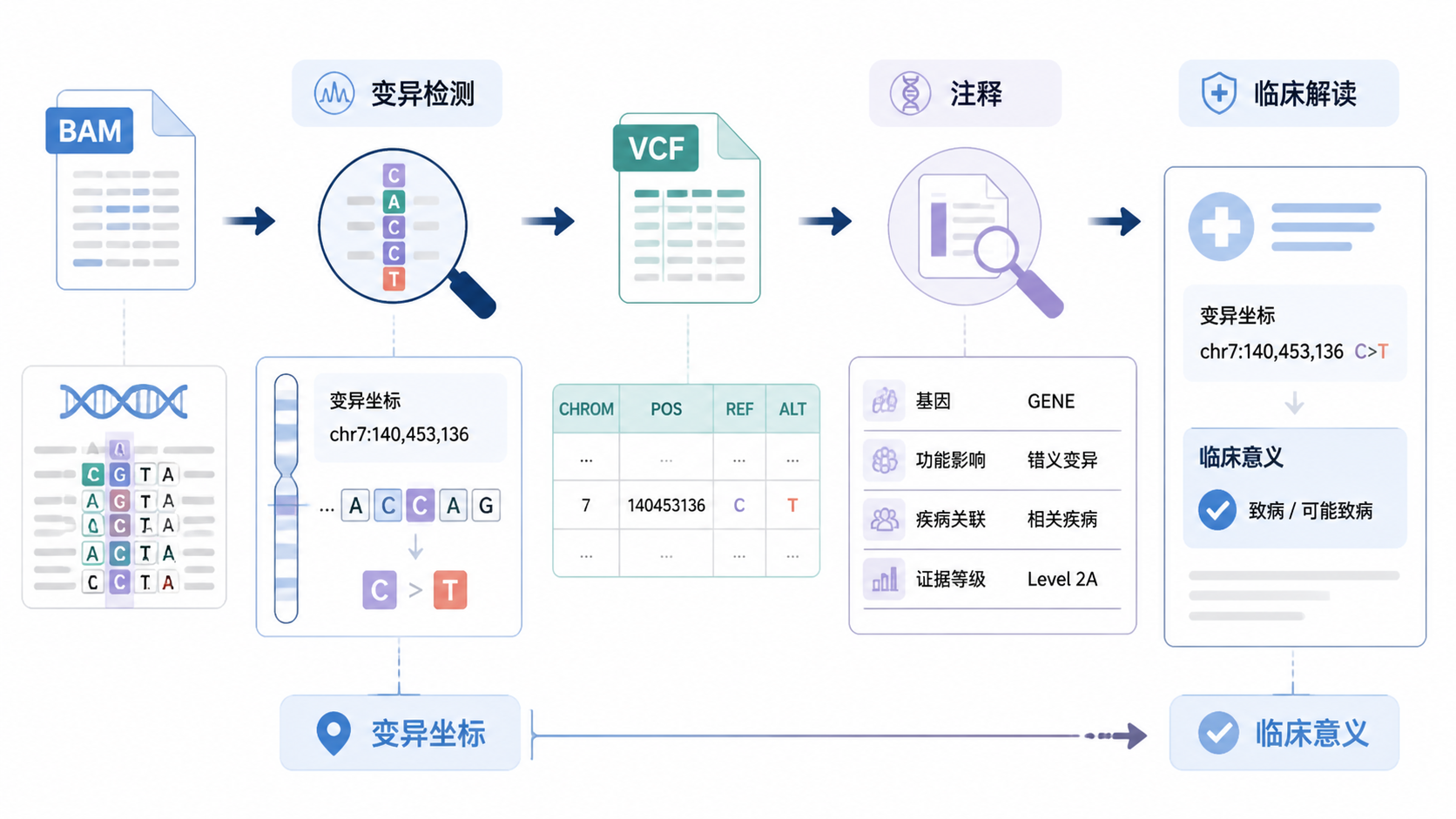
VCF文件注释的价值,就是把原始变异转成可解释、可报告、可决策的信息。
1. 先理解VCF文件注释的输入基础
1.1 VCF不是终点,而是中间产物
VCF文件来自variant calling,常见工具包括GATK、samtools、freebayes、Atlas等。它记录的是变异位点、参考碱基、替代碱基、质量值和过滤标记。
但这还不够。没有注释的VCF,无法区分frameshift、missense、stop gain,也无法判断它是否落在外显子、内含子或UTR。
在实际工作中,VCF文件注释通常建立在前面的质控和过滤之上。比如,变异检测前要看bam文件的coverage、mapping rate和MAPQ。变异检测后,还要看VCF是否通过pass筛选、是否存在clustered mutations、污染、PCR重复和PON可疑位点。
这些步骤决定了最终进入注释的变异是否可信。
1.2 注释前必须明确样本类型
胚系突变和体细胞突变,注释逻辑并不相同。
胚系变异更关注遗传风险、致病性和家族共分离。常结合ClinVar、ClinGen等数据库。
体细胞变异更关注突变丰度、克隆来源和靶向治疗证据等级。常结合药物证据、肿瘤相关数据库和临床指南。
因此,VCF文件注释前,先要明确是生殖系分析,还是肿瘤体细胞分析。否则同一个变异,解释会完全不同。
2. 方法一,基于基因组区域的功能注释
2.1 先回答“这个变异在哪”
这是VCF文件注释的第一层。软件会先判断变异落在哪个区域。常见类别包括:
- 外显子区。
- 内含子区。
- UTR5和UTR3。
- 上游区域。
- 基因间区。
区域注释决定了这个变异是否可能直接影响蛋白编码。
例如,外显子区的变异更容易产生frameshift、missense、synonymous、stop gain等结果。
而内含子或上游区域的变异,往往需要进一步结合剪接位点、调控元件和已知数据库证据来解释。
2.2 再回答“它可能造成什么后果”
区域确认后,下一步是功能后果分类。常见结果包括:
- synonymous。
- missense。
- nonsense或stop gain。
- stop loss。
- frameshift insertion。
- frameshift deletion。
- inframe insertion和inframe deletion。
这是VCF文件注释里最基础,也最常被临床阅读的一层。
其中,frameshift通常提示读码框改变,影响较大。
stop gain会导致提前终止。
stop loss可能导致蛋白延长。
这类标签在报告中非常重要,因为它们直接关系到蛋白功能是否受损。
3. 方法二,基于转录本的标准化注释
3.1 一个变异可能对应多个转录本
同一个基因常有多个转录本。一个变异可以同时落在不同转录本上,注释结果也会不同。
这就是为什么VCF文件注释不能只看基因名,还要看参考转录本。
有些团队会用最长转录本,有些会使用LRG推荐转录本。两者各有优缺点。
临床场景下,关键是要尽量选到最有助于患者解释和治疗决策的转录本。
3.2 不同软件对转录本处理不完全一致
VEP、Annovar、SnpEff都能做转录本注释,但输出格式和排序逻辑不同。
VEP注释更丰富,更新快,维护团队大。Annovar文档成熟,使用广。SnpEff也常用于基础分析。
转录本选择不统一,是VCF文件注释中最容易造成结果差异的来源之一。
因此,报告中必须明确使用了哪个参考转录本体系,避免同一变异出现不同解释。
4. 方法三,基于数据库的致病性和人群频率注释
4.1 用数据库判断“见过没有,常不常见”
数据库注释是VCF文件注释的核心方法之一。
人群频率数据库可以帮助过滤常见良性变异,减少假阳性解释。常用思路是查看变异在人群中是否高频出现。
如果一个变异在人群中频率较高,通常更倾向于germline常见多态性,或者对疾病影响有限。
如果频率很低,尤其在疾病相关基因中,就需要进一步评估其临床意义。
4.2 用临床数据库判断“是否已知致病”
对于胚系变异,常看:
- ClinVar。
- ClinGen。
ClinVar是常用的临床变异数据库。ClinGen的证据整理更严格。
致病性通常分为五级:
- 致病。
- 可能致病。
- 意义未明。
- 可能良性。
- 良性。
这一步是VCF文件注释从“生物信息结果”走向“临床解读”的关键。
对于肿瘤相关基因,还要结合基因本身的疾病谱。比如BRCA1、BRCA2、TP53、CDH1、POLE等,分别对应不同肿瘤易感或驱动背景。
数据库证据能帮助判断这个变异更偏向遗传易感,还是肿瘤体细胞事件。
5. 方法四,基于体细胞临床证据等级的注释
5.1 体细胞变异必须看治疗意义
在肿瘤场景中,VCF文件注释不能只停留在“这是个突变”。
还要回答:它有没有药物价值,有没有入组价值,有没有指南支持。
临床上常按证据等级分层。可概括为:
- A级,FDA已批准或权威指南明确推荐。
- B级,高质量临床研究或专家共识支持。
- C级,临床研究或入组标准支持。
- D级,主要来自临床前研究或早期证据。
这类分层非常适合肿瘤体细胞VCF文件注释。
因为同样一个变异,可能只在少数患者中有治疗价值。证据等级能帮助医生判断是否进入进一步验证或治疗讨论。
5.2 变异丰度也必须纳入注释框架
体细胞分析里,突变丰度很重要。它与肿瘤细胞比例、亚克隆比例、样本纯度有关。
血液样本中,还要考虑ctDNA在cfDNA中的占比。
没有丰度背景的VCF文件注释,临床解释往往不完整。
一个低丰度突变,可能是真实亚克隆事件,也可能只是测序噪音。
因此,注释报告应结合VAF、深度和样本背景一起看。
6. 方法五,基于自动化注释软件的标准化整合
6.1 三大常用注释软件
目前常用的VCF文件注释软件主要有三类:
- VEP。
- Annovar。
- SnpEff。
VEP由Ensembl团队维护,更新快,注释能力强。
Annovar在学术界使用广,文档完整。
SnpEff也可完成基础注释,但维护和生态相对弱一些。
软件的意义,不只是“跑出结果”,而是把区域、转录本、功能后果、数据库证据统一整合。
6.2 为什么自动化很重要
一份WES报告可能达到一两百页。手工整理变异注释,既慢又容易出错。
自动化流程可以把VCF文件注释、筛选、分类和报告输出串联起来,提高效率和一致性。
对于临床和科研团队来说,自动化带来的好处很直接:
- 减少人工抄录错误。
- 提高批量样本处理效率。
- 统一注释标准。
- 方便后续复核和追踪。
7. 做好VCF文件注释,重点不是“多”,而是“准”
VCF文件注释常见问题,不是数据库不够多,而是信息太杂。
真正有价值的结果,应当满足三个条件:
- 位置明确。
- 功能后果明确。
- 临床证据明确。
只有把区域、转录本、数据库和证据等级串起来,VCF文件注释才有临床意义。
对于科研人员,这决定了后续功能验证的优先级。
对于医生,这决定了是否需要遗传咨询、是否需要进一步检测。
对于医学生,这也是理解肿瘤基因检测报告的核心入口。
总结Conclusion
VCF文件注释的5大核心方法,可以概括为区域功能注释、转录本标准化注释、数据库致病性注释、体细胞证据等级注释,以及自动化软件整合注释。它们共同完成了从变异坐标到临床意义的转换。
如果没有这一步,VCF只是文件;完成这一步,VCF才成为可解释的医学证据。
在实际工作中,建议优先建立稳定、可复核、可追溯的注释流程。这样才能在WES和肿瘤分子诊断中,既保证速度,也保证准确性。
如果你希望把VCF文件注释做得更规范、更高效,可以借助解螺旋的标准化流程和报告体系,减少人工整理负担,让结果输出更稳定。

- 引言Introduction
- 1. 先理解VCF文件注释的输入基础
- 2. 方法一,基于基因组区域的功能注释
- 3. 方法二,基于转录本的标准化注释
- 4. 方法三,基于数据库的致病性和人群频率注释
- 5. 方法四,基于体细胞临床证据等级的注释
- 6. 方法五,基于自动化注释软件的标准化整合
- 7. 做好VCF文件注释,重点不是“多”,而是“准”
- 总结Conclusion