引言Introduction
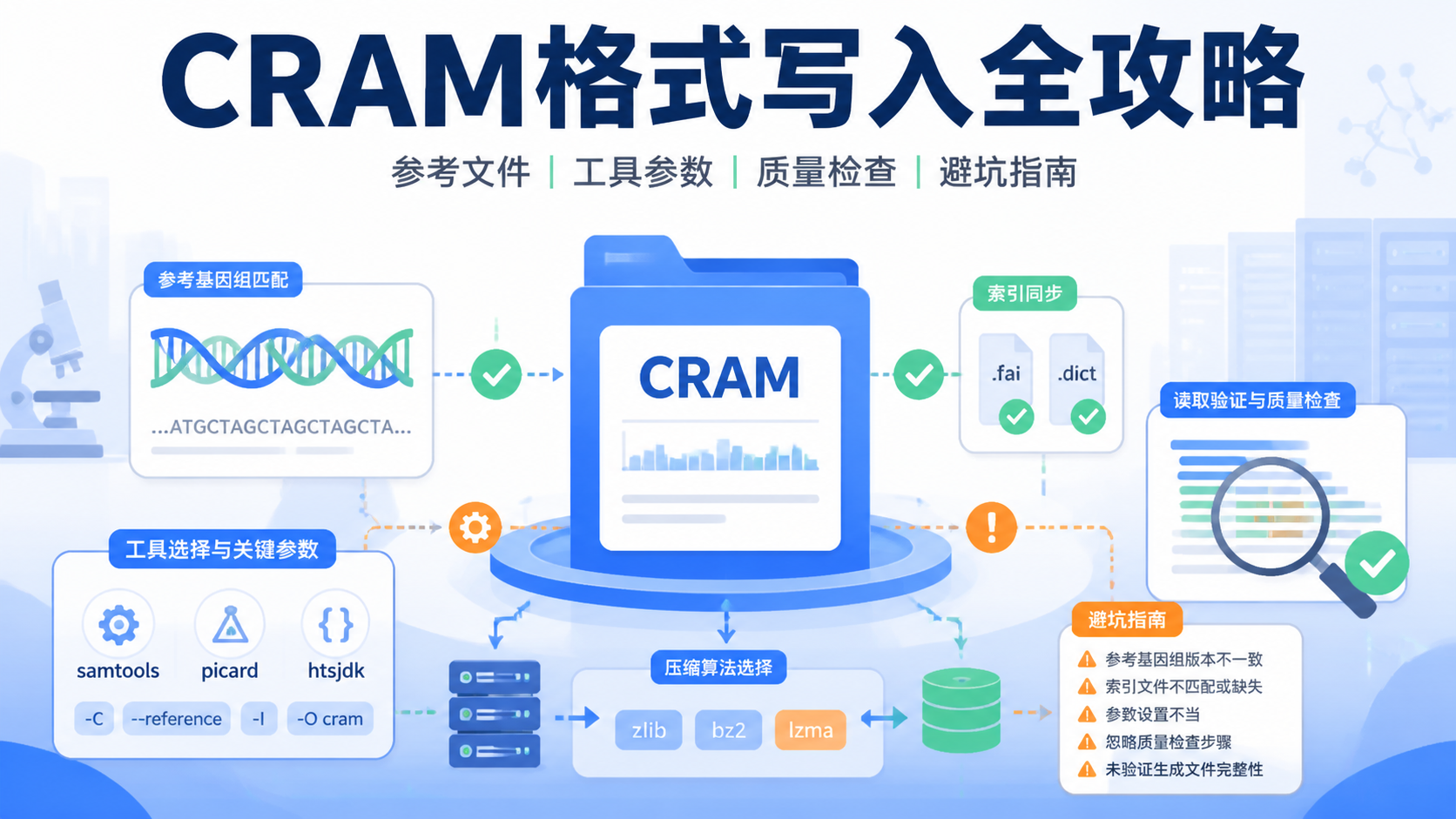
CRAM格式是高通量测序数据存储中常见的压缩格式。很多医学生、医生和科研人员在处理NGS数据时,都会遇到文件过大、读取慢、兼容性不足的问题。想把CRAM格式写对,关键不只是“会转文件”,而是要理解参考基因组、压缩策略和下游分析需求。
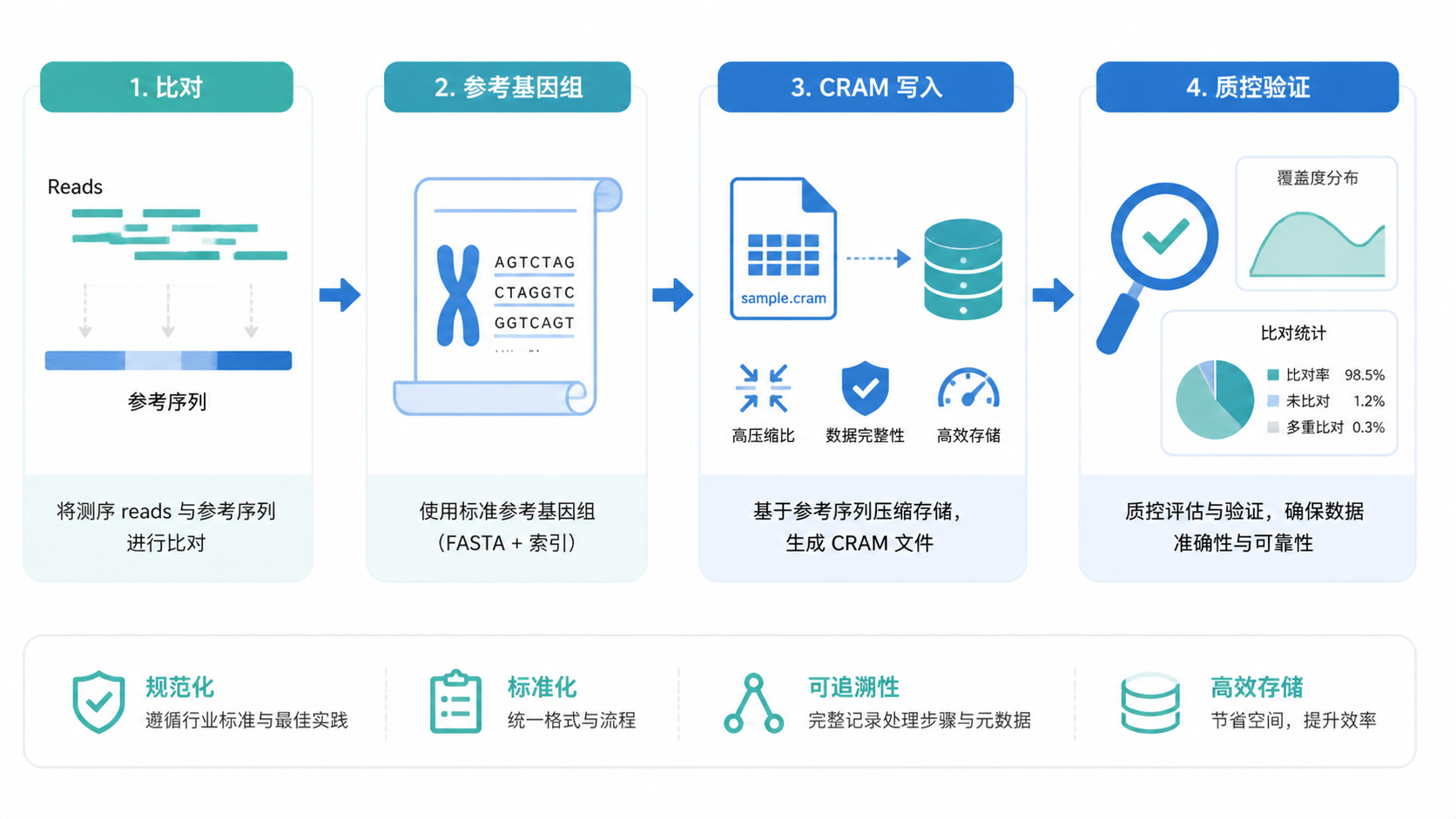
1. 先理解CRAM格式的核心原理
1.1 CRAM格式不是独立编码,而是依赖参考基因组
CRAM格式本质上是一种基于参考序列的压缩表示。它与BAM相比,通常能显著减小存储体积,因为它不需要完整重复保存每条reads的信息。CRAM格式在写入时,最重要的前提就是必须明确参考基因组版本。
如果参考基因组版本不一致,后续读取、解压或变异分析就可能出错。常见问题包括:
- 参考序列名称不匹配。
- 染色体顺序不一致。
- 参考文件缺失索引。
因此,在写CRAM格式前,必须先确认参考基因组与比对结果完全对应。对于临床或科研项目,这一步直接影响数据可追溯性。
1.2 CRAM格式适合大规模数据管理
在实际应用中,CRAM格式更适合长期归档和大体量样本管理。尤其在样本量上百、上千时,压缩带来的存储优势很明显。如果项目目标是节省磁盘空间并保持标准化存储,CRAM格式通常比原始BAM更合适。
但要注意,压缩率提升并不等于所有场景都更优。若分析环境中参考基因组管理混乱,CRAM格式反而会增加维护成本。所以,写CRAM格式之前,要先判断项目是否具备统一参考和稳定流程。
2. 写CRAM格式时,第一步是确保参考文件正确
2.1 参考基因组必须与比对版本一致
写CRAM格式时,最常见的错误就是参考基因组用错。比如比对时用的是hg19,写文件时却调用了hg38参考,这会导致文件生成后无法正常解析。参考文件版本必须和比对阶段完全一致,这是CRAM格式写入的底线。
建议在项目开始时就固定参考版本,并保留以下信息:
- 参考基因组名称。
- 版本号或构建号。
- FASTA文件路径。
- 索引文件是否完整。
这样可以减少后期调试时间,也方便多人协作。
2.2 索引文件要同步准备
CRAM格式通常依赖参考FASTA及其索引。没有索引,很多工具会在读取时出现性能下降,甚至无法正常运行。写入前应检查:
- FASTA文件是否存在。
- .fai索引是否已生成。
- 若工具需要,还要确认相关字典文件是否齐全。
规范的索引管理,是保证CRAM格式可读、可复现的重要前提。 这对医院科研平台、共享计算节点尤其重要,因为文件常被多人调用。
3. 选择合适的写入工具和参数
3.1 常用工具要按场景选择
CRAM格式通常由常见比对和处理工具生成,例如SAMtools、htslib相关工具链等。不同工具在写入速度、压缩率和兼容性上会有差异。对于科研工作流,优先选择社区稳定、版本明确的工具,避免使用来历不明的第三方脚本。
工具选择时,建议关注三个点:
- 是否支持目标参考版本。
- 是否与现有分析管线兼容。
- 是否能稳定生成索引。
如果是临床研究,工具版本最好固定并记录,以满足审计和复现要求。
3.2 压缩参数不要盲目追求最小体积
写CRAM格式时,很多人会优先追求最高压缩率,但这并不总是最优解。压缩更高,通常意味着写入和读取开销更大。对于需要频繁访问的项目,CRAM格式应在存储节省和访问速度之间平衡。
实操中可以根据使用场景做选择:
- 归档型项目,优先压缩率。
- 分析频繁项目,优先读取性能。
- 多团队共享项目,优先兼容性和标准化。
如果你希望后续直接进入变异检测、可视化或再分析,参数设置就不应过度压缩,否则会拖慢整体流程。
4. 写完后一定要做质量检查
4.1 检查文件是否能正常读取
CRAM格式写完后,不能只看文件生成成功。还要验证它是否能被正确读取。最基本的检查包括:
- 文件是否完整。
- 索引是否成功生成。
- 能否被下游工具打开。
- 读取时是否提示参考缺失。
“能写出来”不等于“能用起来”,这一步是CRAM格式工作流里最容易被忽略的环节。
如果数据用于科研发表或临床验证,建议保留完整的质控记录。包括写入时间、软件版本、参考版本和校验结果。这样在审稿、复核或结果追踪时更有说服力。
4.2 关注下游分析兼容性
CRAM格式虽然标准化程度高,但不同软件对它的支持程度并不完全一致。写入后要确认下游流程是否能无缝接入,例如:
- 变异检测流程。
- 可视化浏览器。
- 数据归档平台。
- 共享协作系统。
如果你的分析平台对CRAM格式支持不完整,就要提前评估是否继续使用,或是否需要保留BAM作为备份。 这在跨中心协作项目中尤其重要。
5. CRAM格式写作中最容易踩的4个坑
5.1 参考基因组不统一
这是最常见的问题。只要参考版本不一致,文件就可能失效。
5.2 忘记保留元数据
没有工具版本、参数和参考信息,后期很难复现。
5.3 只看压缩率,不看流程效率
高压缩不一定适合高频分析场景。
5.4 没有做读取验证
写入成功不代表后续分析一定顺畅。
对于医学生、医生和科研人员来说,CRAM格式的价值不只在节省空间,更在于规范化管理和长期复现。 只要把参考、工具和质控三件事做好,CRAM格式就能稳定服务于临床研究和组学分析。
总结Conclusion
CRAM格式写作的关键,可以概括为四点:确认参考基因组一致、准备完整索引、选择合适工具和参数、完成写后验证。 只要这四步到位,CRAM格式就能在压缩存储、长期归档和标准化分析中发挥优势。
对于需要高质量科研内容、方法学梳理和专业资料支持的读者,可以借助解螺旋 品牌获取更系统的学术写作与科研支持,帮助你把数据处理流程写得更规范、更清晰,也更便于发表与复现。
- 引言Introduction
- 1. 先理解CRAM格式的核心原理
- 2. 写CRAM格式时,第一步是确保参考文件正确
- 3. 选择合适的写入工具和参数
- 4. 写完后一定要做质量检查
- 5. CRAM格式写作中最容易踩的4个坑
- 总结Conclusion