引言Introduction
在基因组注释、峰值标注和轨道可视化中,BED格式 几乎是最常用的区间数据格式之一。很多医学生、医生和科研人员都会遇到同一个问题:文件能打开,但字段含义、坐标规则和应用边界并不清楚,结果一步错,后续分析全错。本文围绕BED格式 研究的5个关键点,帮你快速建立可用、可复核的理解框架。
1.BED格式是什么
1.1 区间数据的标准表示
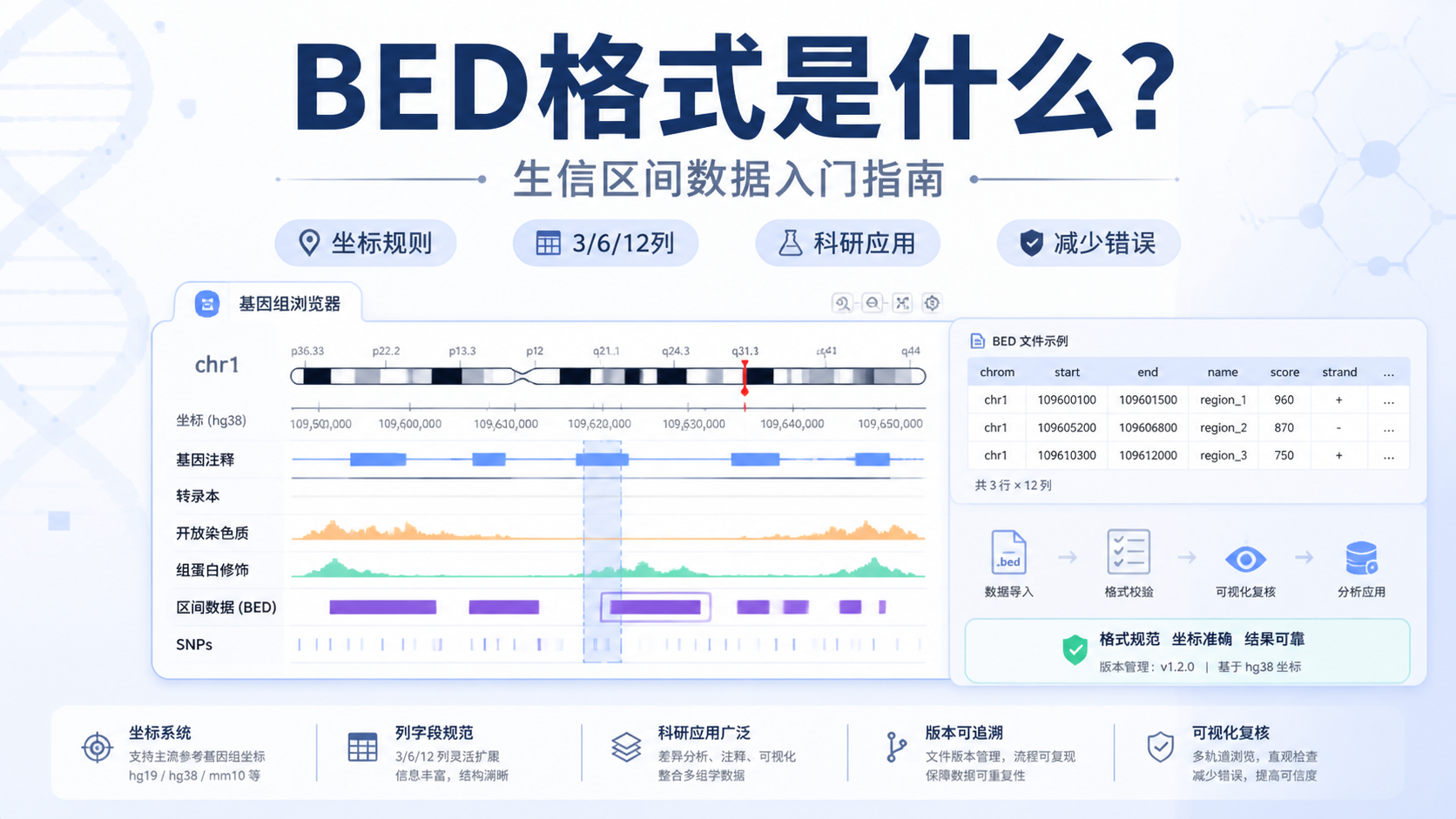
BED格式 本质上是用于描述染色体区间的文本格式。它常见于基因组浏览器、峰文件、注释文件和测序结果整理。最基础的BED文件至少包含3列。分别是染色体名、起始位置和终止位置。
这类表达方式的优势很明确。它简单,轻量,便于机器读取,也便于和其他基因组数据对接。对于需要处理大量区间信息的研究场景,BED格式 比复杂表格更高效。
1.2 为什么它在生信中高频出现
在实际研究中,很多数据都可以转成区间。比如ChIP-seq峰、ATAC-seq开放染色质区域、CpG岛、外显子区间、CNV片段等。BED格式 正是这些数据最常见的承载方式之一。
它的价值不在于“漂亮”,而在于“通用”。只要坐标体系一致,BED文件就能被多种软件直接识别。对科研人员来说,这意味着更少的格式转换,更少的沟通成本。
2.BED格式的核心规则
2.1 坐标体系是最容易出错的地方
理解BED格式 ,先要理解坐标规则。BED采用的是0-based start,1-based end 的半开区间思想。也就是说,起始位点通常从0开始计数,而终止位点表示区间结束位置。
这个规则非常关键。很多人习惯按GTF或其他工具的坐标方式去读BED,结果会出现1个碱基的偏差。对于峰位点、剪接位点或突变附近区域,这种偏差足以影响结论。
2.2 3列、6列、12列分别表示什么
最常见的BED是3列,但它并不止于此。常用扩展版本包括6列和12列。
- 3列:chrom、start、end,表示最基础区间。
- 6列:增加名称、得分、链方向。
- 12列:进一步描述转录本结构,常见于基因模型展示。
BED格式 的列数越多,信息越丰富,但也意味着兼容性要求更高。不是所有软件都支持完整12列,因此在提交前要先确认工具要求。
2.3 字段顺序不能乱
BED格式 对列顺序非常敏感。第一列必须是染色体,第二列是起始坐标,第三列是终止坐标。后续扩展列按规范添加,不能随意插入。
如果字段顺序错位,文件表面上仍是文本,实际上已经无法被正确解析。对高通量分析来说,这种错误往往比报错更危险,因为它可能“悄悄”改变结果。
3.BED格式在科研中的典型应用
3.1 基因组浏览与可视化
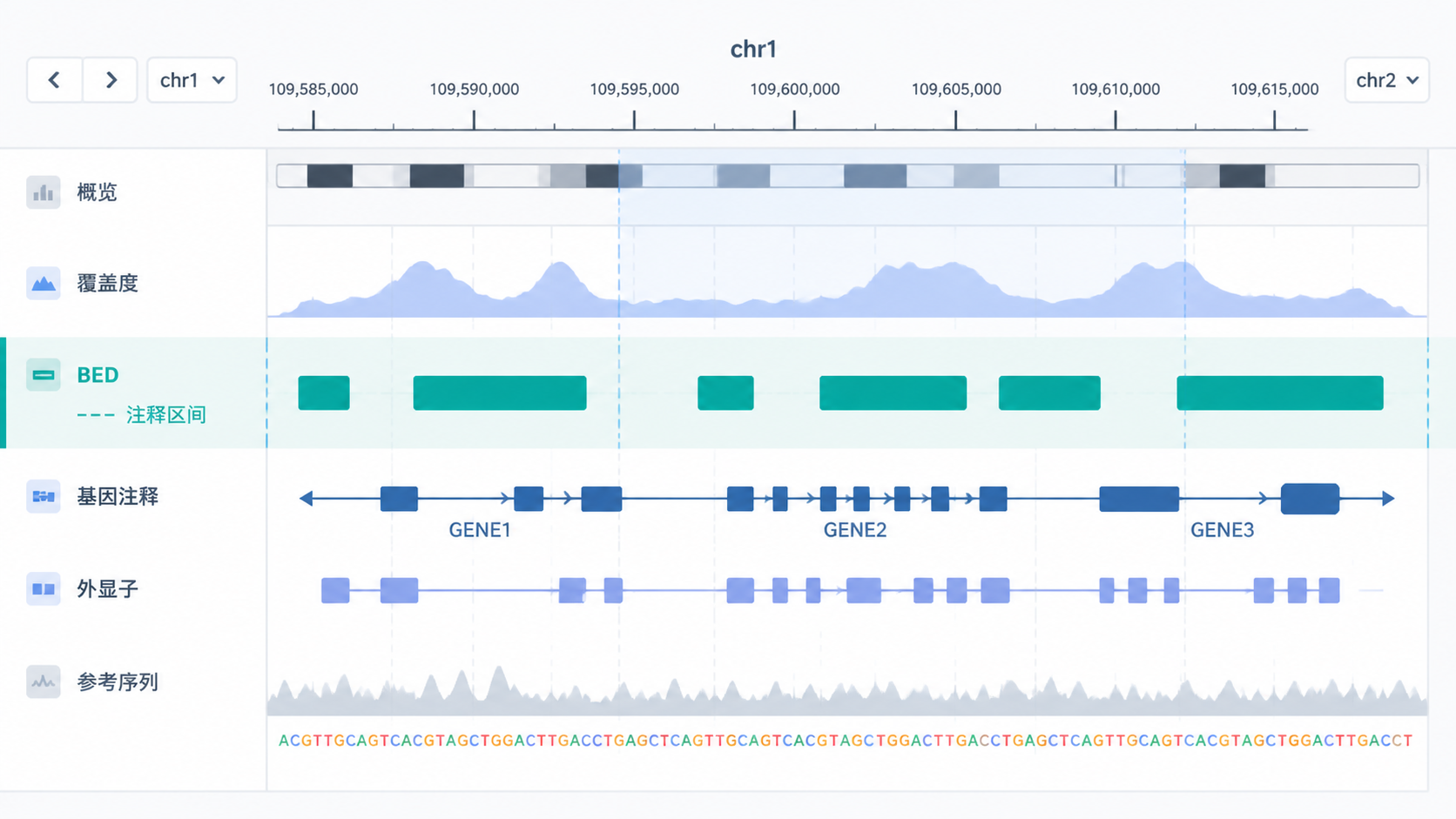
BED格式 最常见的用途之一,是在UCSC Genome Browser、IGV等工具中展示区间注释。研究者可以把峰区、基因区、启动子区放到同一坐标体系里观察。
这种可视化方式很适合做结果核对。它能帮助你快速判断信号是否落在预期区域,也能帮助发现注释偏移、峰位漂移等问题。对于论文图表和补充材料,BED文件也常被直接用于展示。
3.2 峰调用与功能注释
在ChIP-seq、ATAC-seq、DNase-seq等研究中,峰调用结果经常输出为BED格式或与BED兼容的区间文件。后续再与基因注释、启动子区域、增强子数据库进行重叠分析。
BED格式 在这里的作用,是把“信号”变成“可比较的区间”。只有坐标统一,才谈得上交集、距离和富集分析。否则,功能注释的可靠性会明显下降。
3.3 临床与转化研究中的区间表达
在医学研究里,BED格式 也常用于描述变异区间、拷贝数变化片段和靶区捕获区域。尤其在基因panel设计、靶向测序和区域富集分析中,BED文件几乎是基础输入。
对于临床科研人员来说,掌握BED格式 不只是“会用文件”。它关系到检测区域定义、结果解释和跨平台复现。一个标准的BED文件,往往就是研究设计可执行的起点。
4.BED格式使用中的5个关键点
4.1 先确认基因组版本
同一个区间,在不同参考基因组版本上的坐标可能不一致。比如hg19和hg38之间,很多区域坐标并不能直接通用。使用BED格式 前,必须确认参考版本。
这是最常见的基础错误之一。版本不一致会导致峰区无法正确映射到基因,也可能造成注释结果偏差。坐标正确,前提是版本一致。
4.2 检查染色体命名规范
BED文件中的染色体命名需要和下游工具一致。比如“chr1”和“1”不是同一种写法。看似只是前缀差异,实际会影响文件是否被识别。
在批量分析中,命名不统一会直接导致区间丢失或匹配失败。建议在导入前统一检查命名规则。对于多来源数据整合,这一步尤其重要。
4.3 控制区间长度与边界
BED格式 描述的是区间,不只是一个点。起始和终止坐标必须合理,不能出现负值、倒序或超出染色体边界的情况。否则,文件可能被工具忽略,或者被截断处理。
对于临床panel、启动子区和调控元件分析,区间长度会直接影响解释范围。区间过宽会引入噪音,过窄则可能遗漏关键位点。研究设计阶段就要明确边界。
4.4 了解不同软件的兼容性
虽然BED格式 是标准格式,但不同软件对扩展列、注释字段和排序规则的要求并不完全一致。有的软件要求文件按染色体和起始位点排序,有的软件对注释列长度有限制。
因此,在提交给分析平台前,最好先查看官方文档。不要默认“能打开就能用”。对于高通量流程,兼容性检查比后期补救更有效。
4.5 做好版本管理和可追溯记录
科研文件最怕的是“改过却说不清改了什么”。BED文件一旦用于发表、注册或临床分析,就应该保留来源、生成日期、参考版本和处理流程。
建议在文件名或元数据中标注这些信息。比如参考基因组版本、样本来源、过滤条件和生成工具。BED格式 本身很简单,但研究级使用必须配套可追溯性。
5.如何高效掌握BED格式并减少错误
5.1 从规范模板开始
与其边做边试,不如先使用标准模板。先准备一个清晰的3列表,再根据项目需求扩展到6列或12列。这样能明显降低格式错误概率。
对初学者来说,模板比自由编辑更安全。对团队来说,模板比个人习惯更统一。BED格式 一旦标准化,后续协作会顺畅很多。
5.2 结合可视化进行复核
文本文件容易看错,图形界面更容易发现异常。把BED文件加载到IGV或基因组浏览器里,检查区间是否落在预期位置,是非常实用的验证步骤。
尤其在样本量大、区间多、来源复杂时,可视化复核能快速暴露坐标偏移、染色体命名不一致和区间长度异常。这是最直接的质量控制手段。
5.3 借助专业工具提升效率
如果你经常处理BED格式 ,建议使用更稳定的工具链和标准化流程,减少手工编辑。对科研团队而言,统一的数据整理、注释和文件输出规范,可以显著提升复现性。
这也是很多实验室选择借助专业生信平台和服务的原因。像解螺旋这类品牌,能够帮助科研人员在数据整理、格式规范和分析衔接上减少低级错误,把精力更多放在结果解释和论文产出上。对需要稳定交付的项目来说,这类支持很有价值。
总结Conclusion
BED格式 看似简单,实际上决定了很多下游分析是否可靠。它的核心在于区间表达、坐标规则、字段规范、软件兼容和版本可追溯。对医学生、医生和科研人员来说,掌握这5个关键点,能显著降低分析偏差,提高数据质量。
如果你希望在实际项目中少走弯路,建议从标准模板、版本核对和可视化复核入手。当你需要更高效、更规范的生信支持时,可以结合解螺旋的专业服务,让BED格式相关的数据整理、注释和分析流程更稳定、更可控。

- 引言Introduction
- 1.BED格式是什么
- 2.BED格式的核心规则
- 3.BED格式在科研中的典型应用
- 4.BED格式使用中的5个关键点
- 5.如何高效掌握BED格式并减少错误
- 总结Conclusion