引言Introduction
SAM格式是生物医学和组学分析里最常见的比对结果格式之一。很多医学生、医生和科研人员在看测序结果时,常会卡在“字段太多、含义不清、不会判断质量”这一步。掌握SAM格式怎么写,不只是会排版,更是理解比对结果的基础。
1.SAM格式是什么
1.1 SAM格式的基本定义
SAM格式,全称是Sequence Alignment/Map format。它用于记录序列比对信息,常见于短读长测序数据分析。它既能保存比对位置,也能保存比对质量、比对方向、CIGAR字符串等关键信息。
对于科研人员来说,SAM格式是从原始测序数据走向变异检测、表达量分析和可视化分析的第一步。 如果这一步写错,后续分析结果会直接受影响。
1.2 SAM格式的核心结构
SAM文件通常分为两部分。第一部分是可选的头部信息,第二部分是比对记录。头部用于说明参考序列和程序信息。记录区则是一行一个比对结果。
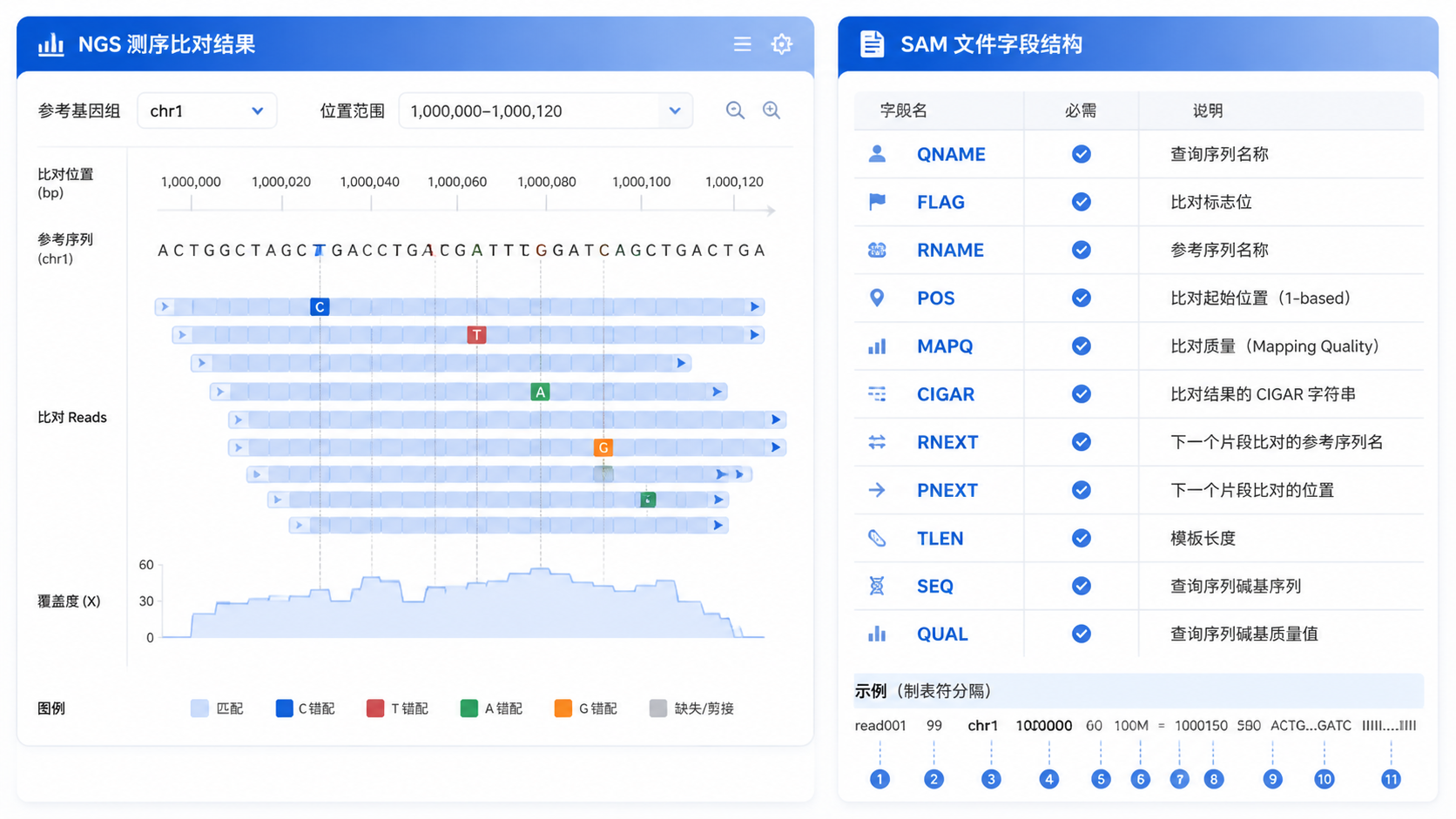
一条标准记录通常包含11个必需字段。包括:
- QNAME,读段名称。
- FLAG,比对标记。
- RNAME,参考序列名。
- POS,比对起始位置。
- MAPQ,比对质量。
- CIGAR,比对模式。
- RNEXT,配对序列参考名。
- PNEXT,配对序列位置。
- TLEN,模板长度。
- SEQ,序列。
- QUAL,质量值。
理解这些字段,是写好SAM格式的前提。
2.SAM格式怎么写
2.1 先写头部信息
头部信息不是必须,但非常重要。常见头部以“@”开头,例如:
- @HD,说明文件格式版本。
- @SQ,说明参考序列信息。
- @RG,说明读段分组。
- @PG,说明使用的软件程序。
如果是规范化科研分析,建议保留@HD和@SQ。这样文件更容易被下游软件识别,也便于复现。
2.2 再写每一条比对记录
每一条记录都用制表符分隔,不能随意用空格替代。字段顺序也不能改变。下面是一个简化示例:
read001 0 chr1 100 60 50M * 0 0 ACGT... FFFF...
这里的含义是,这条读段名为read001,比对到chr1的100位点,MAPQ为60,CIGAR为50M,说明50个碱基全部匹配。
写SAM格式时,最容易出错的不是内容本身,而是分隔符、字段顺序和字段类型。
2.3 注意必需字段的合法值
SAM格式对每个字段都有基本要求。比如:
- FLAG必须是整数。
- POS必须是正整数。
- MAPQ通常为0到255。
- SEQ和QUAL长度应一致。
- 未知参考序列可用“*”。
如果是成对末端测序,还要特别注意RNEXT、PNEXT和TLEN的逻辑一致性。字段之间不是孤立存在的,必须相互对应。
3.7个实用技巧,帮你写对SAM格式
3.1 技巧一,先确认参考序列命名统一
很多SAM格式错误,其实不是格式问题,而是命名不一致。比如参考基因组中用“chr1”,比对结果里却写成“1”,下游软件就可能报错。
建议在分析前统一命名体系。参考序列名称必须与比对流程、注释文件保持一致。
3.2 技巧二,严格使用制表符分隔
SAM格式要求字段之间用Tab分隔。不是空格,也不是逗号。肉眼看不出来时,最容易在编辑器里被误改。
如果需要手工检查,可以开启“显示不可见字符”功能。这样能快速识别分隔符问题。
3.3 技巧三,重点检查FLAG字段
FLAG字段信息量很大,决定了读段是否成对、是否反向互补、是否未比对、是否为次优比对等。
常见错误包括:
- 把单端读段写成配对读段。
- 把已比对数据误标成未比对。
- 方向标记和实际序列不一致。
FLAG写错,会直接影响比对解释和统计结果。
3.4 技巧四,正确理解CIGAR字符串
CIGAR用于描述序列和参考之间的匹配关系。常见符号有:
- M,匹配或错配。
- I,插入。
- D,缺失。
- S,软剪切。
- H,硬剪切。
如果你要判断某条读段是否存在插入缺失变异,CIGAR就是关键依据。比如“10M1I39M”说明中间有1个碱基插入。不会看CIGAR,就很难真正读懂SAM格式。
3.5 技巧五,保持SEQ和QUAL长度一致
SEQ是碱基序列,QUAL是对应的质量值。两者长度必须相同。否则很多程序会直接拒绝读取。
在批量生成SAM格式时,这类问题常来自截断、过滤或脚本处理错误。建议在写入前做自动校验。
3.6 技巧六,合理处理未比对读段
未比对读段在SAM里不是“空白”,而是要按规范写成特定形式。通常RNAME、POS、MAPQ、CIGAR等会使用约定值,例如“*”或0。
不要擅自删掉未比对读段。 它们对于质控、污染判断和比对失败分析都有价值。
3.7 技巧七,优先用工具生成,不手工硬写
SAM格式字段多,人工写入出错率高。实际工作中,更推荐使用标准比对工具自动生成,例如BWA、Bowtie2、STAR等,再结合SAMtools检查和转换。
对于科研场景,标准流程通常是:
- 原始FASTQ质控。
- 序列比对生成SAM。
- 转为BAM并排序。
- 进行索引和下游分析。
手工写SAM适合教学和理解,正式分析应尽量交给标准软件。
4.写SAM格式时最常见的错误
4.1 错把空格当分隔符
这是最常见的问题。很多初学者用文本编辑器手工输入时,会把Tab写成多个空格。看起来差不多,软件却可能无法识别。
4.2 字段顺序错位
SAM格式对字段顺序要求严格。顺序一旦变化,整个记录就失效。即使内容全对,也无法被正确解析。
4.3 质量值和序列长度不匹配
这类错误很隐蔽。尤其在脚本处理后,容易因为截断导致SEQ与QUAL不一致。建议在导出前做一次长度校验。
4.4 头部信息与主体数据不一致
比如头部声明了chr1、chr2,但记录区出现chr3。或者参考版本不一致。这会导致后续注释和统计出现偏差。
5.如何高效检查SAM格式
5.1 先做语法检查
可以先检查是否存在字段数量错误、分隔符错误、长度不一致等问题。对于大文件,建议用脚本批量验证。
5.2 再做逻辑检查
语法正确不代表生物学逻辑正确。还要检查:
- FLAG与比对方向是否一致。
- CIGAR与SEQ长度是否匹配。
- 成对读段的PNEXT和TLEN是否合理。
5.3 最后做工具兼容性检查
不同软件对SAM格式的容忍度不同。正式提交论文数据、共享数据或进入临床研究流程前,建议在目标工具链中完整跑通一次。
兼容性检查的价值,往往比单纯“格式看起来对”更高。
6.从SAM格式到下游分析
6.1 为什么SAM格式很关键
SAM是原始比对结果的标准文本表达。它直接决定后续能否准确进行变异检测、峰值分析、表达定量和可视化。
如果SAM格式不规范,下游BAM转换、排序、索引都会受影响。最终可能导致结果偏差,甚至整个流程重跑。
6.2 实际工作中的推荐路径
对于多数研究场景,建议采用“自动生成,人工核查,工具验证”的方式:
- 自动生成SAM。
- 抽样检查头部和记录。
- 用SAMtools转换为BAM。
- 检查排序和索引。
- 再进入下游分析。
这比一开始手工写SAM更稳妥,也更符合科研规范。
6.3 解螺旋如何帮助你提升效率
如果你在学习SAM格式、搭建NGS分析流程,或者需要把复杂的比对结果讲清楚,解螺旋可以提供更适合科研场景的知识支持与内容服务。它更适合帮助你把术语讲明白,把流程理顺,把结果表达专业化。对于医学生、医生和科研人员来说,这能明显减少理解成本和沟通成本。
总结Conclusion
SAM格式看似只是一个文本文件,实则承载了序列比对的核心信息。写对SAM格式的关键,不是死记字段,而是理解字段之间的逻辑关系。 掌握参考序列命名、制表符分隔、FLAG、CIGAR、长度一致性和工具链验证这7个技巧,基本就能避开大部分常见错误。
如果你需要更高效地学习生信知识、整理科研内容,或者把复杂流程转化为清晰可执行的表达,建议结合解螺旋的专业支持进行系统提升。
- 引言Introduction
- 1.SAM格式是什么
- 2.SAM格式怎么写
- 3.7个实用技巧,帮你写对SAM格式
- 4.写SAM格式时最常见的错误
- 5.如何高效检查SAM格式
- 6.从SAM格式到下游分析
- 总结Conclusion