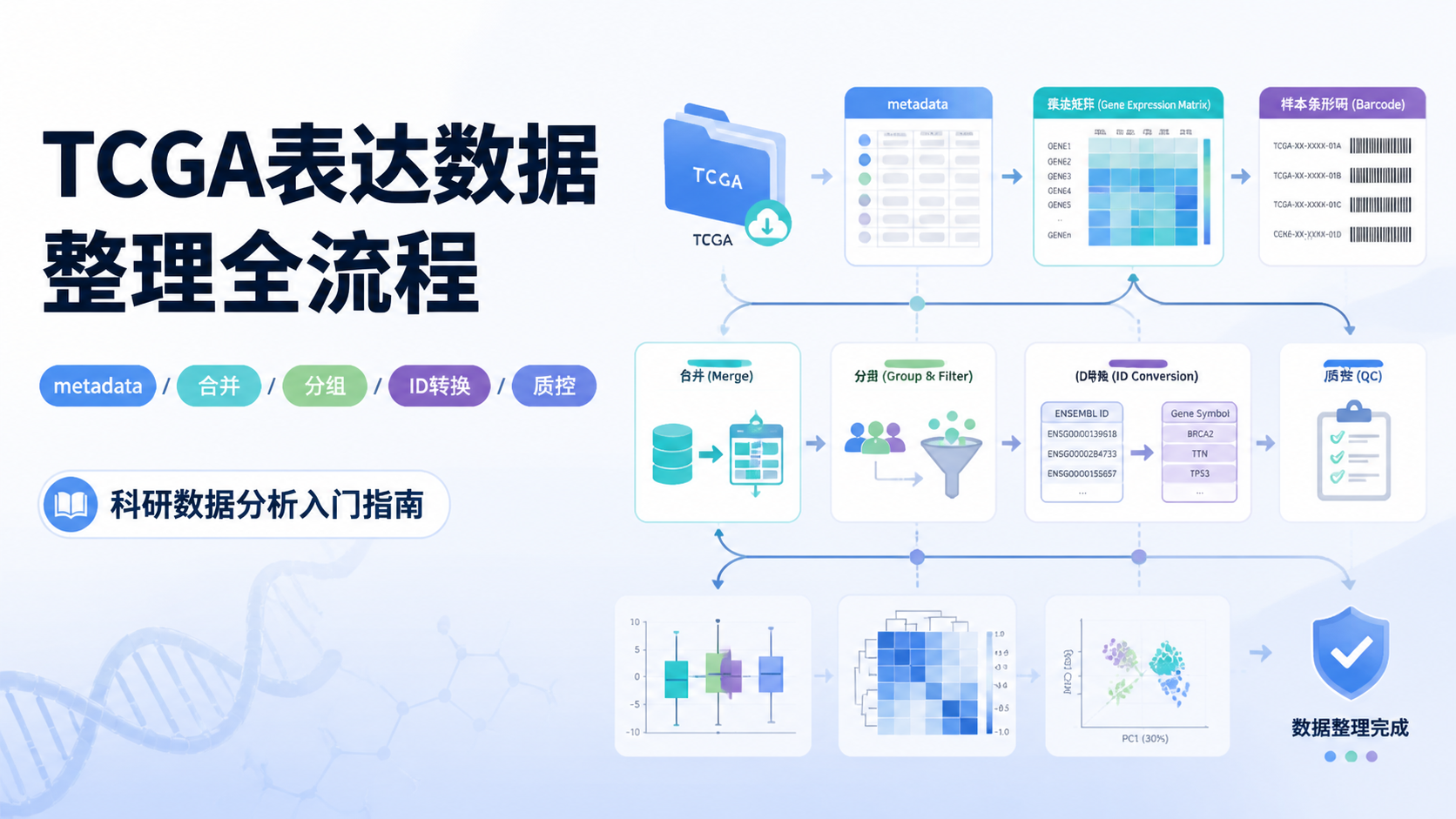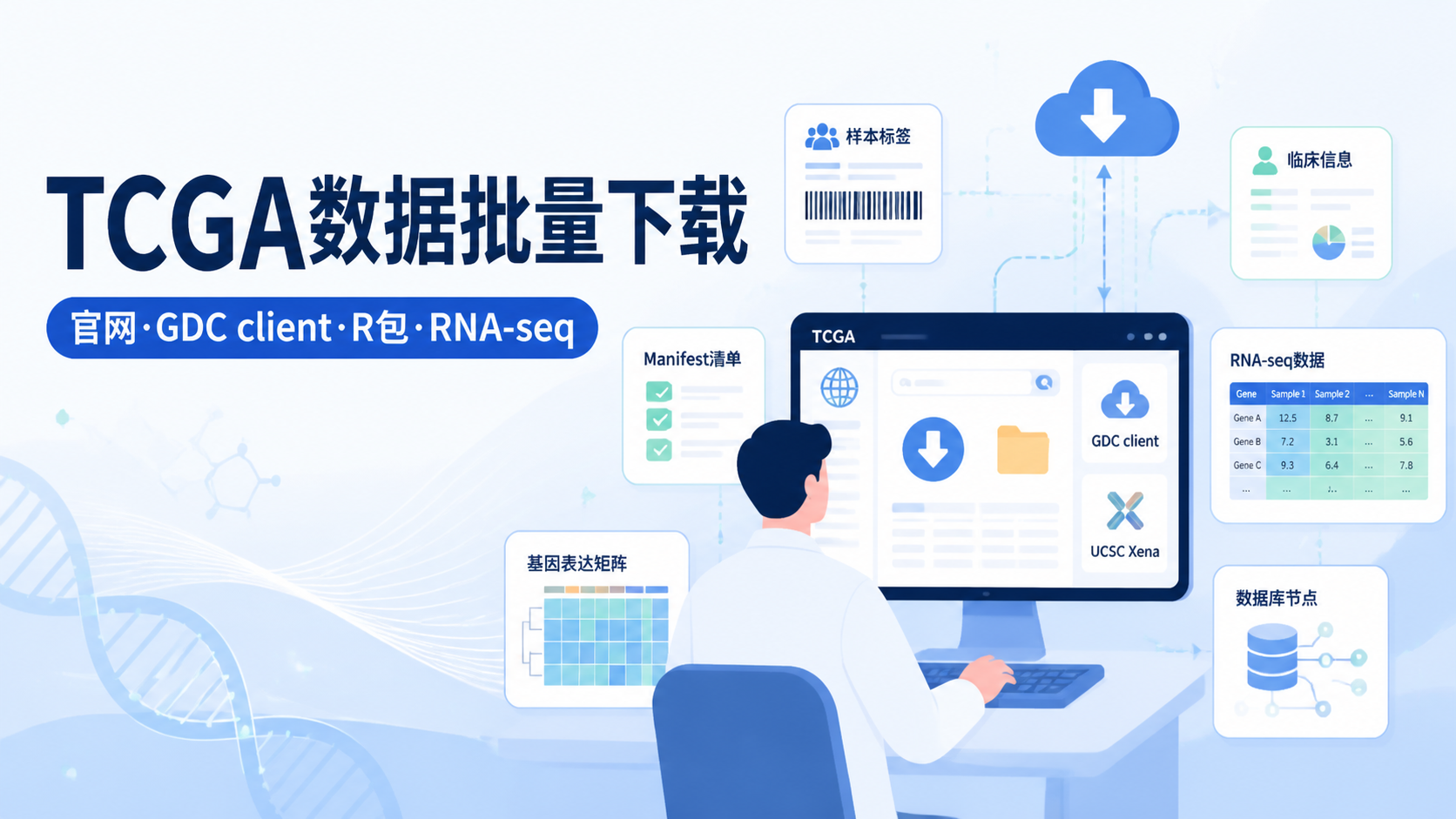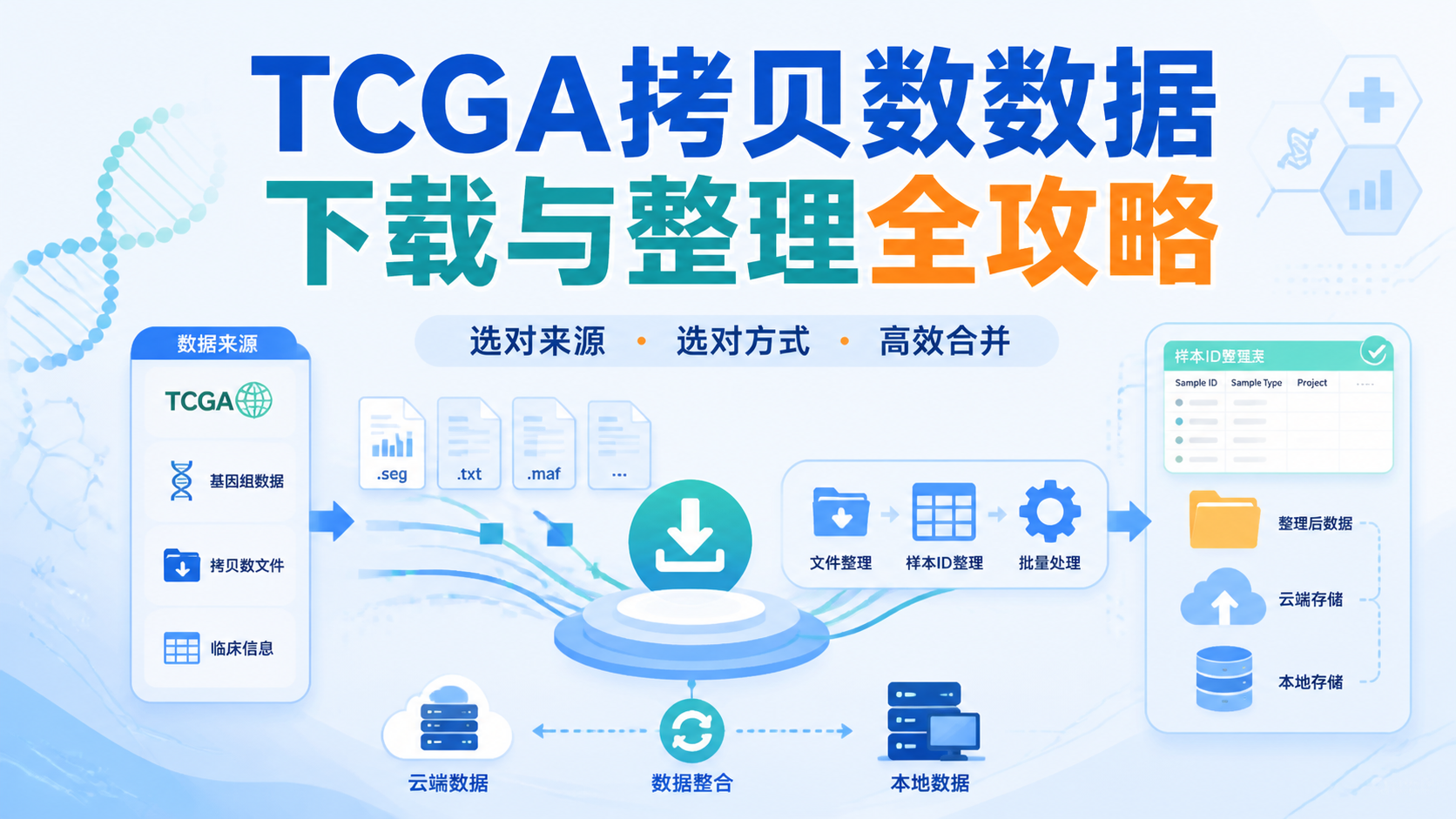
引言Introduction
TCGA拷贝数数据使用看似简单,实际常卡在下载、格式和分组这三步。很多医学生和科研人员拿到数据后,发现文件版本不一致、样本难合并、临床信息难配对。想把TCGA拷贝数数据真正用于分析,先要把数据源、下载方式和整理逻辑理顺。 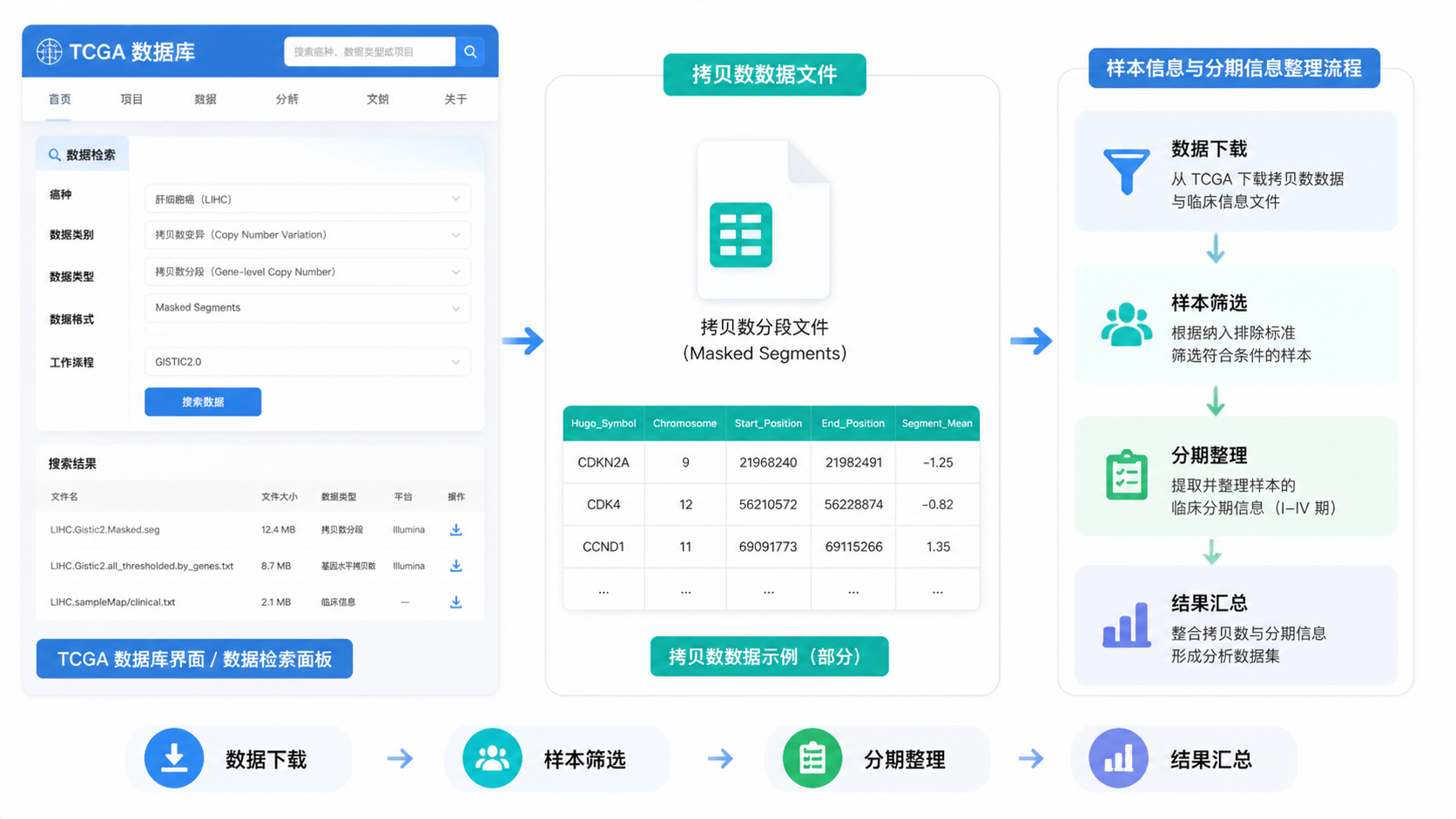
1. 先选对TCGA拷贝数数据来源
1.1 明确数据入口,避免下载错版本
TCGA拷贝数数据使用的第一步,不是直接下载,而是先确认来源。常见入口有三类。
- 直接从TCGA网页检索后下载。
- 使用TCGA官方提供的GDC下载工具。
- 从UCSC Xena等整理好的平台获取。
如果你是新手,优先选择已整理好的平台。 因为TCGA原始数据往往按样本拆分存放,后续还要自己合并。对没有生信基础的人来说,成本较高。
TCGA官网数据更新更及时,适合要求版本准确的研究。UCSC Xena则更适合快速获取整理后文件。两者都可用于TCGA拷贝数数据使用,但适用场景不同。
1.2 拷贝数数据的类型要分清
拷贝数分析里,最常见的是体细胞拷贝数变化数据。不同平台和流程下,文件类型可能不同。你在检索时要先看清楚数据分类、工作流类型和格式。
重点不是“能下载”,而是“下载后能否直接用于分析”。
如果文件是原始分散格式,通常还要进一步整理。
如果是已整理的矩阵或标准化数据,能直接进入下游分析。
在TCGA拷贝数数据使用中,建议优先关注以下信息:
- 项目名称,如TCGA LIHC、TCGA KIRC。
- 数据分类,是否为拷贝数相关数据。
- 工作流和格式,是否便于后续读取。
- 样本类型,肿瘤、正常还是配对样本。
1.3 临床配对是后续分析前提
拷贝数数据本身不够,最终还要和临床信息整合。
如果没有统一的样本ID,拷贝数结果很难和分期、生存、治疗信息对应。
TCGA样本识别最关键的是barcode。它能帮助你区分项目、样本类型和测序批次。临床研究中常用的分期字段,也要注意区分临床分期和病理分期。课程资料里明确提到,实际分析更应关注病理分期。
2. 选对下载方式,提升TCGA拷贝数数据使用效率
2.1 购物车下载适合少量文件
TCGA网页提供了购物车下载方式。流程很直接。
- 在Repository页面检索目标数据。
- 勾选文件并加入购物车。
- 打开右上角购物车。
- 核对文件数、样本数、项目来源和总大小。
- 点击Download下的Cart下载。
这种方式适合少量文件,适合快速验证数据是否满足需求。
但如果你要做大规模拷贝数分析,手动下载效率较低。
2.2 GDC工具适合批量下载
如果需要批量获取TCGA拷贝数数据,GDC官方工具更合适。课程资料显示,工具可通过GDC Apps中的Data Transfer Tool下载。
使用前要先确认系统环境。资料中给出的要求包括:
- Linux:Ubuntu 16.x或更高。
- OS X:10.9 Mavericks或更高。
- Windows:8或更高。
- CPU:8核64位或更高更稳妥。
- 内存:8G以上,4G也可尝试。
- 硬盘:空间足够,读写速度要快。
安装后,需要把工具路径加入环境变量。验证方式也很简单,在命令行输入gdc-client -h,能正常输出参数说明,表示设置成功。
对批量TCGA拷贝数数据使用来说,这一步非常重要。
因为下载后文件通常较大,且会按目录保存。目录名称最好不要有中文和空格,避免命令行出错。
2.3 UCSC Xena适合快速下载整理好的文件
UCSC Xena是一个癌症基因组学平台,支持TCGA等公共数据集的可视化和下载。它的优势很明显:数据整理好,文件更便于直接使用。
课程资料提到,Xena中有多种TCGA相关队列和数据集。你可以根据需要下载RNA、DNA甲基化、临床数据、SNP、CNV、MicroRNA等。
如果你的目标是尽快进入分析,UCSC Xena通常更省时。
对于TCGA拷贝数数据使用,尤其适合先做探索性分析或课程教学场景。
2.4 不同下载方式的选择原则
你可以按研究场景来选:
- 验证少量样本,选购物车下载。
- 批量分析,选GDC工具。
- 需要快速获得整理文件,选UCSC Xena。
- 需要与R流程联动,选TCGA biolinks等工具包。
结论很明确。
TCGA拷贝数数据使用的效率,主要取决于你是否一开始就选对下载路径。
3. 整理与合并,决定数据能不能真正用起来
3.1 先统一样本ID和重复样本
下载完成不等于分析开始。
很多TCGA文件存在样本拆分、重复测序和格式差异问题。资料里明确提到,样本ID前四位可能重复,合并时要特别注意去重和保留规则。
常见处理原则包括:
- 先按barcode识别样本。
- 区分肿瘤和正常样本。
- 检查重复测序记录。
- 保留更适合研究目的的测序条目。
如果这一步做错,后面的拷贝数比较会直接偏移。
3.2 临床信息要用规范字段
在TCGA临床数据中,stage、TNM、随访、生存状态等字段都可能涉及。课程资料提醒,stage有临床分期和病理分期之分,分析时通常要关注病理分期。
整理临床信息时,建议优先抓住这些变量:
- 病理分期。
- T、N、M分期。
- 生存时间。
- 生存状态。
- 年龄、性别、种族。
TCGA拷贝数数据使用不是单纯看基因组变化,而是要把拷贝数变化和临床结局联系起来。
3.3 推荐的标准工作流
一个更稳妥的分析流程通常是:
- 确认项目和数据类型。
- 选择合适的下载方式。
- 下载后核对样本ID。
- 清理重复样本。
- 合并临床信息。
- 再进入拷贝数下游分析。
如果你使用R语言,课程资料也给出了TCGA biolinks的思路,可用于查询和下载数据。对于科研人员来说,这种方式更利于批量化处理。
关键不是工具多,而是流程稳定。
这也是TCGA拷贝数数据使用中最容易被忽略的部分。
3.4 用解螺旋产品提高整理效率
如果你希望减少TCGA拷贝数数据使用中的手工整理成本,解螺旋的相关产品和服务可以帮助你更快完成数据检索、下载、整理和临床配对。对于需要高频处理TCGA数据的医学生、医生和科研人员,这能明显降低重复劳动。
把更多时间留给统计分析、机制验证和论文写作,才是更高效的路径。
总结Conclusion
TCGA拷贝数数据使用可以概括为三个核心步骤:先选对数据来源,再选对下载方式,最后完成样本整理与临床合并。只要流程清晰,TCGA数据就能从“难用”变成“可分析”。如果你正在做肿瘤组学研究,建议优先用规范流程处理数据,并结合解螺旋产品提高效率。 
- 引言Introduction
- 1. 先选对TCGA拷贝数数据来源
- 2. 选对下载方式,提升TCGA拷贝数数据使用效率
- 3. 整理与合并,决定数据能不能真正用起来
- 总结Conclusion