引言Introduction
TCGA数据预处理是差异分析、临床关联和模型构建前最关键的一步。很多项目卡在样本混乱、标准化不一致、低表达基因过多这三类问题上,导致结果不稳、重复性差。本文以TCGA-COAD为例,拆解tcga数据预处理 的5个核心步骤。
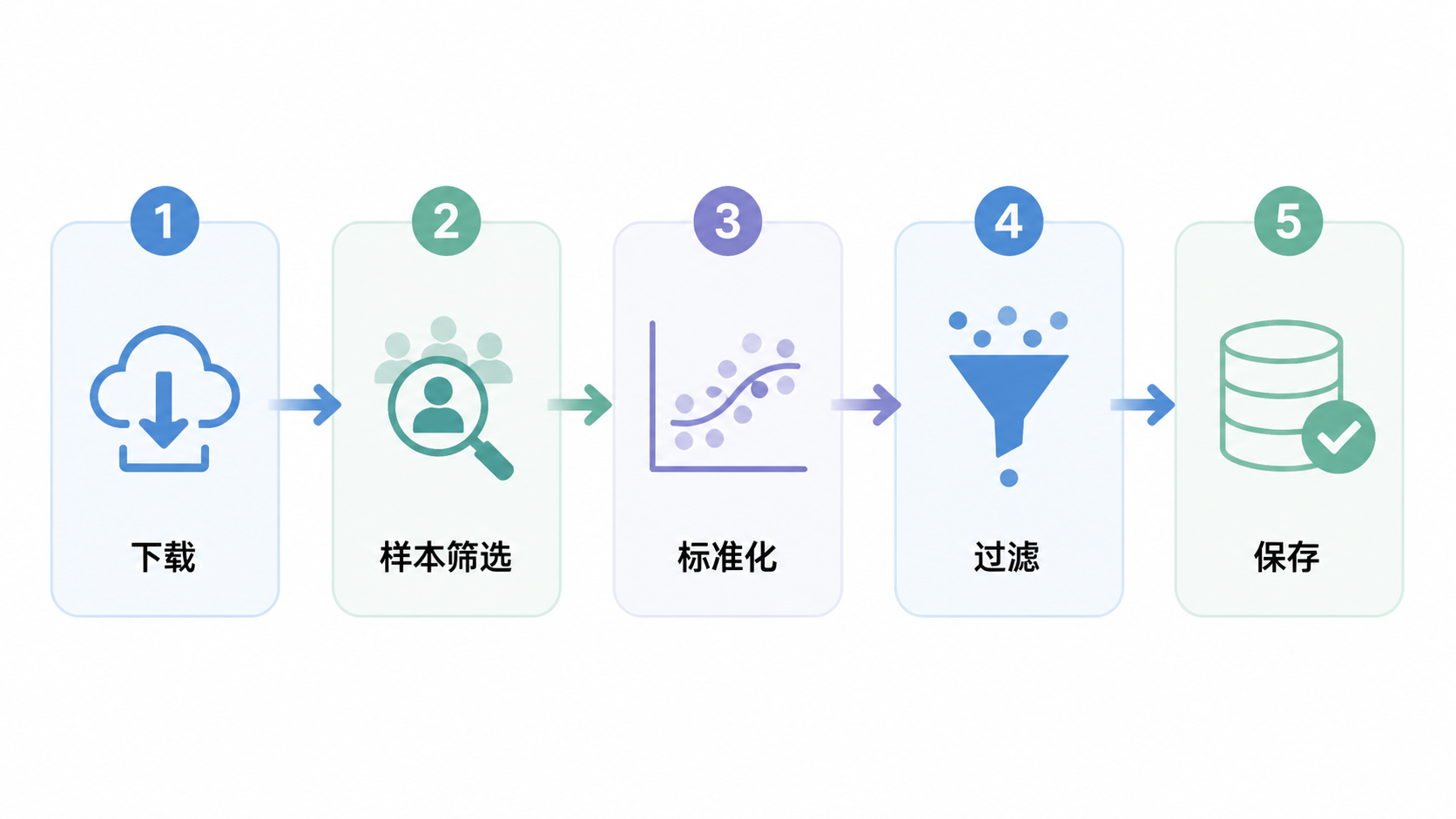
1.TCGA数据下载与样本定位
1.1 先确认项目ID和数据类型
在GDC平台下载TCGA数据时,第一步不是直接点下载,而是先确认项目ID。以结肠癌为例,标准写法是COAD。随后再明确数据类别,如count数据、clinical数据等。这样做的好处很直接,能避免把不相关文件带入分析流程。
tcga数据预处理 的起点,必须建立在正确的数据来源上。课程提纲中明确提到,需要通过project ID找到TCGA COAD的标准写法,并修改代码中的project、data.category等参数后再下载count数据。
1.2 用barcode区分肿瘤和正常样本
TCGA barcode是样本分类的核心依据。根据上游知识库,barcode的第14、15位可以识别样本组织学类型。
- 01,原发肿瘤。
- 11,正常组织。
在TCGA-COAD示例中,样本总数为521个,其中NT样本41个,TP样本478个。先识别样本类型,再进入统计分析,是tcga数据预处理的基本原则。
1.3 先下载再核对样本
实际操作中,通常会先通过getResults获取样本信息,再用barcode参数重新下载特定样本。这样能保证NT和TP样本顺序清楚,后续分组不容易出错。知识库中提到,NT和TP样本会放入c括号中,且NT在前,TP在后,最终保存为TCGA_COAD_count.Rda文件。
2.构建Metadata并核查文件完整性
2.1 Metadata决定样本与文件的一一对应
TCGA数据文件多,直接靠文件名分析风险很高。更稳妥的方法,是先下载JSON格式的metadata,再用R中的jsonlite包读取。Metadata里包含样本名字、样本ID、组织类型等关键信息。
这一步的价值在于,把原始文件名和样本ID准确对应起来。
在课程内容中,metadata会被整理成数据框,提取file name、MD5值、TCGA ID、患者ID和样本类型。这样后续合并表达矩阵时,样本身份不会混淆。
2.2 MD5校验是必要的质量控制
下载完成后,不建议直接进入分析。应先检查文件MD5值是否与metadata一致。知识库明确说明,可以用tools包验证文件完整性。
如果MD5不一致,说明文件可能损坏或下载不完整。对于后续的表达矩阵合并,这类问题会直接导致错误。
tcga数据预处理不是“读进来就能用”,而是先确认数据可信。
2.3 统一整理样本信息
在实际项目里,metadata还常用于提取病例ID、样本类型和批次信息。对于TCGA与GTEx合并场景,这些信息尤其重要,因为它关系到肿瘤与正常样本的比较是否成立。知识库提到,样本注释信息、batch number、TSS编码等,都会影响后续结果解释。
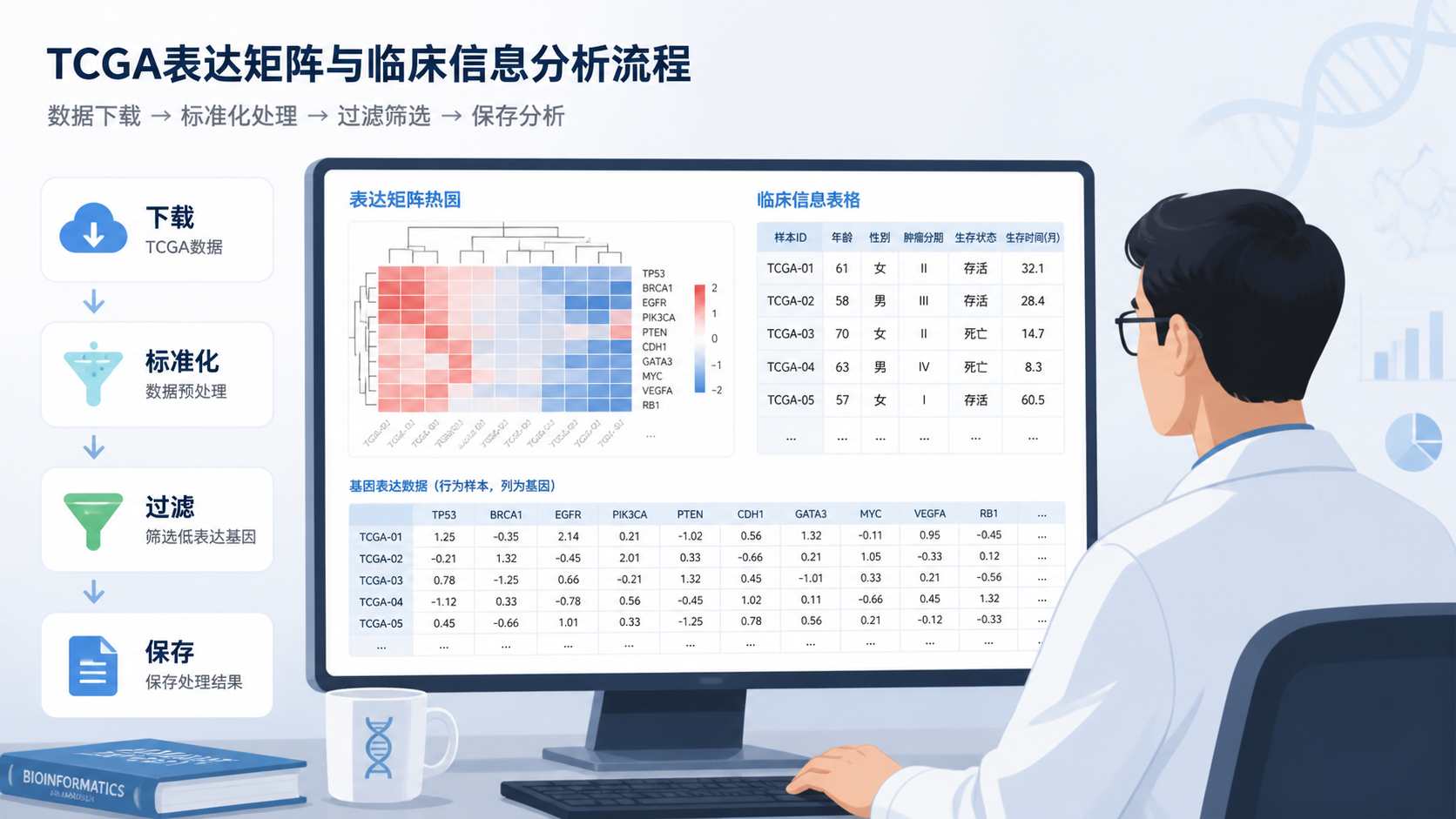
3.读取count矩阵并完成标准化
3.1 先看原始分布,再决定是否标准化
拿到count矩阵后,不要立刻做差异分析。先画箱式图,看样本分布是否存在明显偏移。课程提纲中明确写到,要通过第一个箱式图判断是否需要标准化。
这是一个非常实用的判断步骤。因为如果样本间分布差异明显,直接分析会放大技术噪音。
3.2 使用EDASeq进行标准化
知识库中给出了明确案例:使用EDA seq包进行标准化,分为四步,耗时约367秒。标准化后,数据大小为98.1 MB,基因数量从56602个变为23192个,样本数量为519个。
这说明标准化不仅是数值调整,也会伴随特征整理。标准化的目标,是让样本之间具备可比性。
3.3 标准化后再检查数据结构
标准化完成后,应再次确认矩阵维度和分布是否合理。对于医学生和科研人员来说,这一步很重要,因为它决定后续差异表达分析、聚类分析和生存分析是否可靠。
如果标准化前后样本分布仍异常,建议回头检查样本类型、批次信息和下载参数。
4.按规则过滤低质量样本和低表达基因
4.1 样本过滤先于基因过滤
在TCGA数据中,样本过滤和基因过滤不是同一件事。课程内容强调,要先核查样本annotation信息,再确认病例是否符合研究设计。比如胆管癌和肝癌在样本层面就不能混用。
样本过滤错了,后面所有统计都可能失真。
4.2 基因过滤减少噪音和内存消耗
RNA-seq差异分析前,过滤低表达基因是常规操作。知识库给出的可选标准包括:
- 去除表达量为0的基因。
- 保留至少一半样本中表达量大于0的基因。
- 保留中位数大于0的基因。
- 使用均值或分位数方法过滤。
在TCGA-COAD示例中,采用TCGAbiolinks包的filtering函数,并使用quantile方法过滤均值在0.25以下的基因。过滤后,基因数量从23192个降至17393个,样本数量仍为519个。
这一步能显著减少无信息基因,提高下游分析效率。
4.3 过滤标准要和研究目的一致
不同课题对过滤阈值的要求不同。做差异分析时,可以偏向保守过滤,保留更稳定的基因集合。做机器学习特征筛选时,则需要结合样本量与特征维度平衡。
总之,tcga数据预处理没有唯一模板,但必须有清晰标准,并在方法部分写明。
5.保存结果并进入下游分析
5.1 预处理结束前要做好结果封装
完成标准化和过滤后,不要直接进入建模。应先把结果保存为Rda或其他可复用格式,便于后续差异分析、通路富集和临床关联分析调用。课程提纲中明确提到,需要保存过滤后的数据以便后续差异分析。
5.2 临床数据也要同步清洗
如果研究要做生存分析,还必须同步处理临床数据。知识库中给出的流程包括读取clinical TSV文件、转换days to last follow up为数值、合并days to death和days to last follow up计算OS、将年龄换算成年、标记生存状态、去重并保存。
表达矩阵干净,不代表临床数据也干净。两者必须同步整理。
5.3 用解螺旋产品提高预处理效率
对初学者来说,TCGA数据预处理最耗时的不是写代码,而是理解每一步为什么要做。解螺旋品牌的课程与实操内容,已经把下载、Metadata核查、标准化、过滤和临床清洗串成完整流程,适合医学生、医生和科研人员快速上手。
如果你想减少试错成本,直接跟着成熟流程做,会比零散拼接代码更稳定。 通过解螺旋的实操课程,可以更快建立TCGA分析框架,把时间更多留给结果解释和论文写作。
总结Conclusion
TCGA数据预处理的核心,不是“把数据跑通”,而是让数据在样本、表达和临床层面都可用于严谨分析。标准流程可以概括为5步:下载并定位样本,构建Metadata并核查完整性,完成标准化,执行样本和基因过滤,最后保存并进入下游分析。
只要前处理规范,后续差异分析、分型分析和预后建模的可信度都会明显提高。
如果你正在做TCGA项目,建议直接参考解螺旋的课程体系,把数据预处理、临床清洗和分析流程一次性打通。
- 引言Introduction
- 1.TCGA数据下载与样本定位
- 2.构建Metadata并核查文件完整性
- 3.读取count矩阵并完成标准化
- 4.按规则过滤低质量样本和低表达基因
- 5.保存结果并进入下游分析
- 总结Conclusion