引言Introduction
FASTQ质控是RNA-seq、WES等测序分析的第一道门槛。很多项目失败,不是因为实验设计差,而是因为原始数据没有先做可靠筛查。只有先确认数据质量,后续比对、定量和变异分析才有意义。
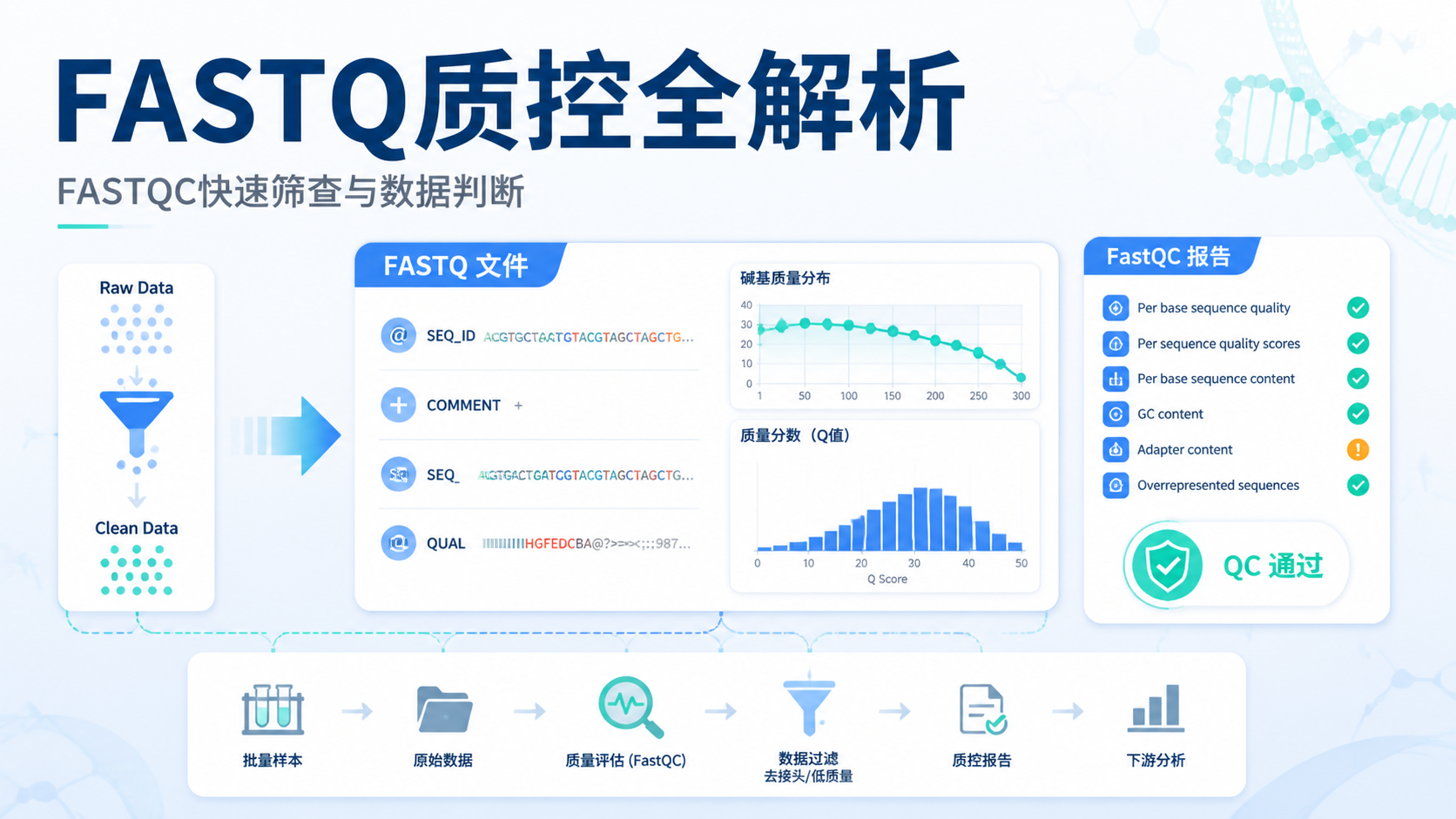
1.FASTQ质控到底在检查什么
1.1 从raw data到clean data的关键一步
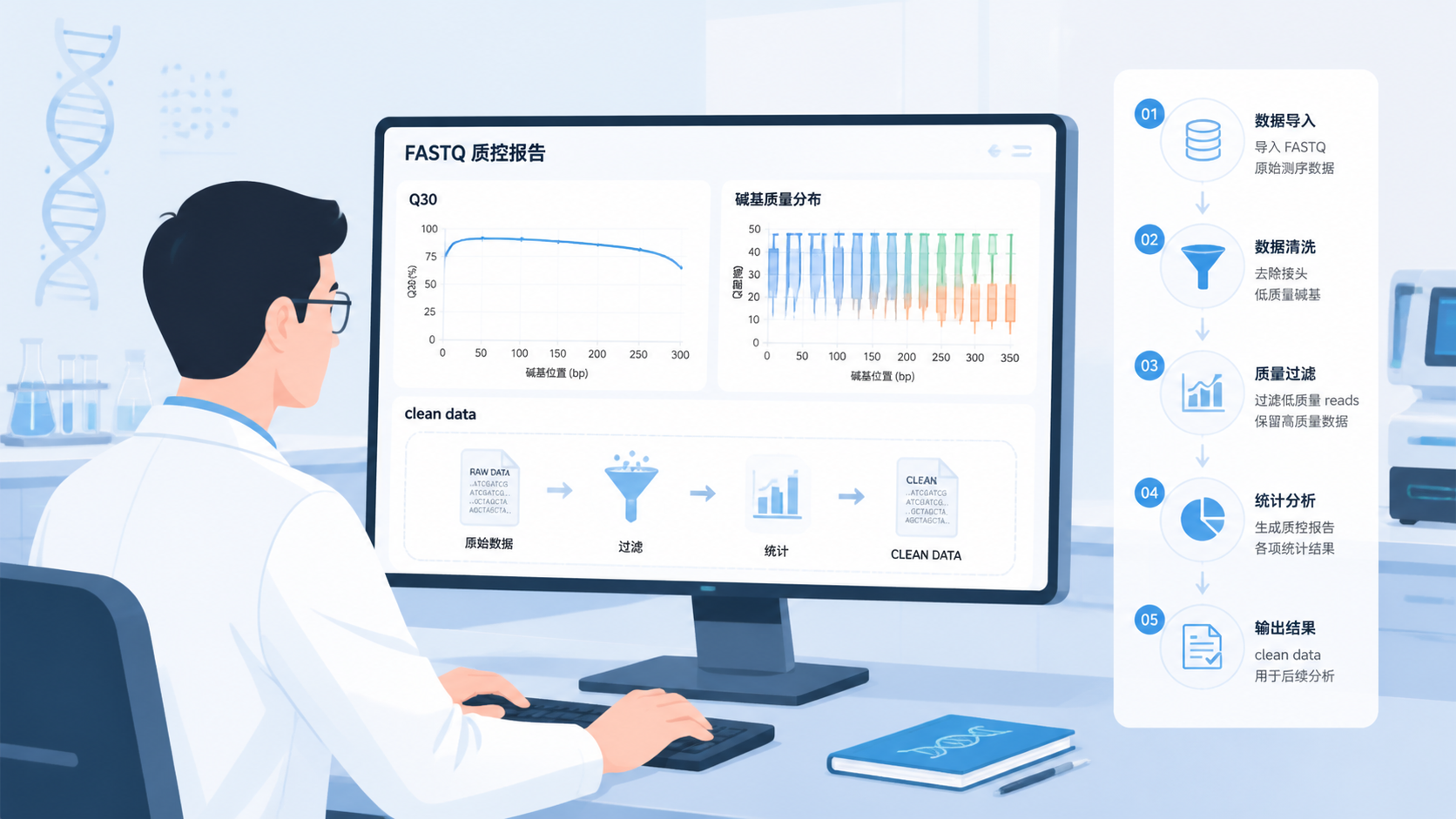
FASTQ质控的核心,是对fastq文件进行剪辑质量值检测,判断测序数据是否可用。这个过程通常包含QC和filter两个环节。前者看整体质量,后者去掉低质量序列,最终得到clean data。
没有通过FASTQ质控的数据,不建议直接进入比对、组装、定量或差异分析。
因为错误会在后续步骤中被放大,影响mapping rate、coverage,甚至导致假阳性和假阴性。
1.2 FASTQ文件的四行结构要先看懂
FASTQ文件每条read有四行,理解这个结构很重要。
- 第一行,以“@”开头,包含序列识别符。
- 第二行,是实际测序序列,通常由ATCG组成。
- 第三行,通常是“+”号。
- 第四行,是与第二行一一对应的质量值。
其中,第二行如果出现N,通常表示该位点测不准。第四行的质量值,直接决定这条read是否值得保留。
1.3 Q值怎么看才算合格
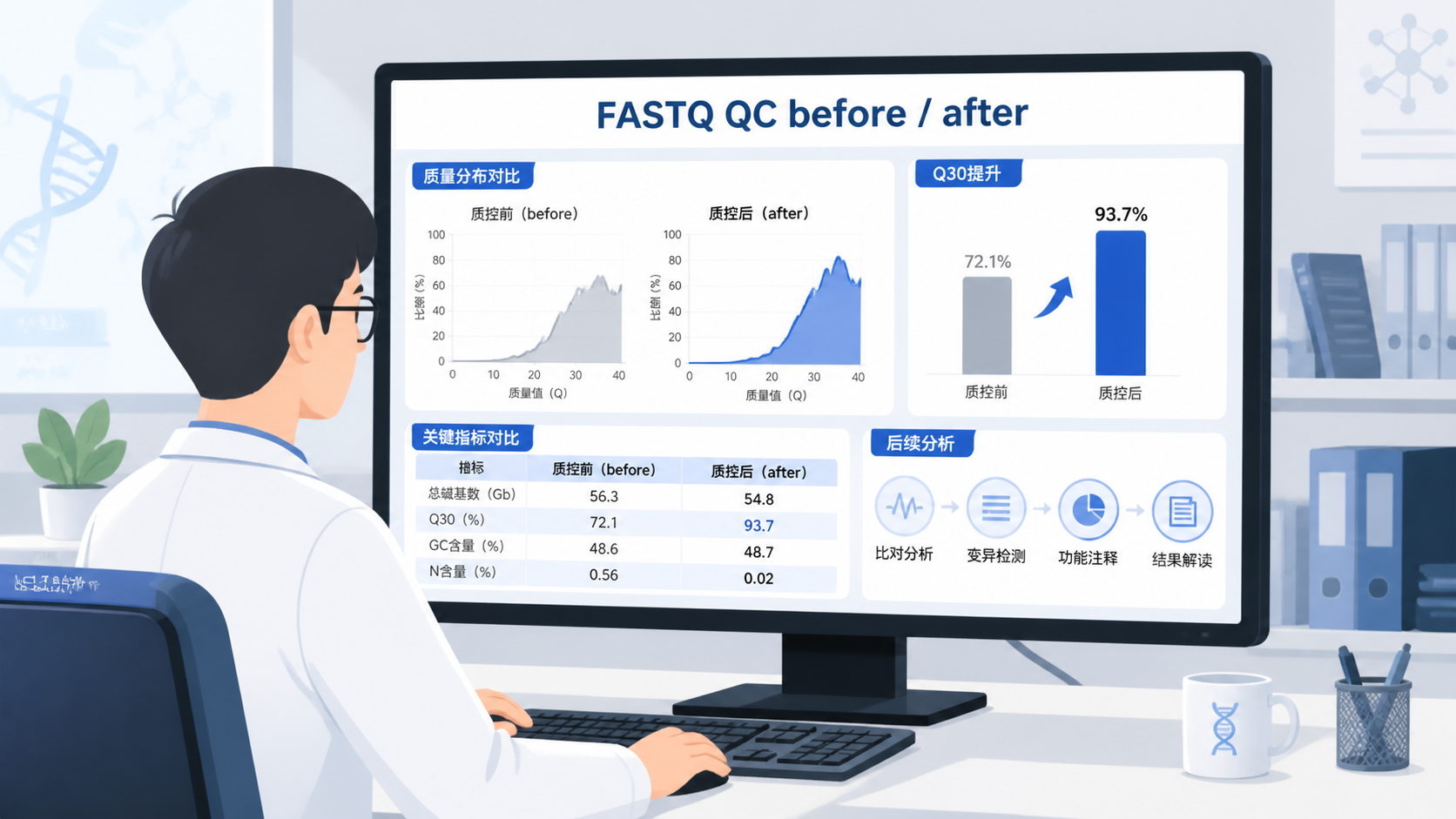
质控里最常见的标准是Q值。课程中明确提到,Q30代表准确率99.9%,Q20代表准确率99%以上。
对于大多数高通量测序项目,重点不是追求每一个碱基都完美,而是看绝大多数碱基是否稳定达到Q30以上。
Q值越高,说明测序错误率越低,后续分析越可靠。
2.如何用FASTQC快速完成基础筛查
2.1 FASTQC是最常用的起点
在实际操作中,FASTQC是最常见的FASTQ质控工具之一。它可以同时处理多个文件,也支持设置输出目录。
常见命令思路很简单:
- 对单个文件做质控。
- 对成对测序文件分别检查。
- 通过-o指定输出路径。
- 通过-t设置线程数,提高运行速度。
对于多个样本,批量质控比逐个手工检查更高效,也更不容易遗漏问题。
2.2 报告里优先看哪些指标
FASTQC报告内容很多,但真正需要优先关注的指标并不多。
建议先看以下几项:
- 碱基质量分布,是否整体位于Q30以上。
- 文件基本信息,如文件名、编码方式、序列长度。
- GC含量,是否明显偏离样本预期。
- Adapter污染,是否提示接头残留。
- 序列长度分布,是否符合建库设计。
其中,碱基质量是最核心的指标。 课程中也强调,绿色区域通常代表通过,说明该部分质量合格。
2.3 不要被某些“常见未通过项”误导
FASTQC里有些项目经常会显示不通过,但并不一定代表数据不能用。
例如:
- sequence duplication levels,重复水平偏高很常见。
- Adapter提示,部分建库类型中并不罕见。
- n碱基含量和长度分布异常,需要结合具体样本判断。
质控不是机械看红绿灯,而是结合测序类型和研究目标判断。
例如,高表达基因本来就会被重复测到多次,因此重复度高不一定是坏事。
3.如何通过质控结果判断数据能不能继续分析
3.1 先看碱基质量,再看整体通过率
FASTQ质控完成后,第一步永远是看碱基质量曲线。只要大部分碱基位于Q30以上,通常就具备进入后续分析的基础。
如果发现以下情况,就要提高警惕:
- 读长末端质量明显下降。
- 某一段区间整体偏低。
- N比例异常升高。
- Adapter污染较重。
这些问题会影响比对效率,也会拉低clean data质量。质控的目标不是“把报告做出来”,而是尽量保住高可信数据。
3.2 GC含量和序列长度要结合样本类型判断
课程中提到,人的GC含量通常大约在50%左右,48%或49%也常见。
因此,GC含量轻微波动往往是正常的。
序列长度分布也一样。不同文库类型、不同测序平台、不同项目设计,都会影响分布形态。
只有明显偏离预期时,才值得进一步排查。
3.3 质控不只是“看结果”,还要看来源
测序技术已经很成熟,Illumina平台也较为稳定,但这不代表可以省略质控。
尤其对主要精力不在数据分析上的课题组来说,QC验证是防止拿到异常数据的重要保险。
建议在项目开始阶段就确认:
- 原始数据是否完整。
- 是否已从SCI或.gz格式正确解压。
- 是否保留了成对文件。
- 是否存在命名混乱或样本混淆。
这些基础问题,会直接影响FASTQ质控效率。
4.批量质控怎么做才更高效
4.1 多样本项目必须批量处理
在真实科研场景中,样本往往不是一个,而是一批。
这时最实用的方法,是用循环命令批量调用FASTQC。
这样做的好处有三点:
- 减少重复操作。
- 降低人为漏检风险。
- 便于统一保存报告。
课程中提到的思路是用while read ID的方式批量运行,再配合线程参数加速。对医学生和科研人员来说,这类批处理方式非常适合常规项目管理。
4.2 线程数和输出路径要提前规划
FASTQ质控时,-o参数控制输出路径,-t参数控制线程数。
这两个参数看似简单,但对效率影响很大。
建议在开始前就确认:
- 电脑或服务器可用线程数。
- 输出目录是否规范。
- 是否按样本名分类保存结果。
这样做可以避免结果文件散乱,后期汇总更轻松。
如果项目样本较多,规范化命名和路径管理,往往比单纯“跑得快”更重要。
4.3 质控只是第一关,后面还有比对
FASTQ质控完成后,下一步才是比对。
也就是说,FASTQ质控并不是终点,而是高质量分析流程的起点。
一个标准的流程通常是:
- FASTQ质控。
- 过滤低质量reads。
- 获得clean data。
- 进入比对、定量、组装或变异检测。
- 再做下游结果解释。
只有前面的数据基础扎实,后面的生物学结论才更可信。
5.为什么FASTQ质控能直接影响项目成败
5.1 低质量数据会放大下游偏差
如果FASTQ质控不充分,低质量碱基、接头污染和异常重复都会进入后续流程。
结果往往是:
- 比对率下降。
- 误差率升高。
- 差异表达或变异检测不稳定。
- 重复实验成本增加。
这也是为什么很多文章和项目,第一步就要求严格查看FASTQ质控报告。
数据质量越早把关,后面返工越少。
5.2 质控结果也能反向提示实验环节问题
FASTQ质控不只是判断“能不能用”,还可以帮助发现实验问题。
例如:
- Adapter异常,可能提示建库或接头残留处理不足。
- 某一端质量明显差,可能提示测序读长末端衰减。
- 重复度异常偏高,可能与文库复杂度不足有关。
这些信息对优化后续实验非常有价值。
对科研人员来说,FASTQ质控也是一个质量反馈工具。
总结Conclusion
FASTQ质控的价值,不在于多看一个报告,而在于尽早识别风险,保住真正可用的数据。掌握FASTQ文件结构、Q值标准、FASTQC报告解读和批量处理方法,就能显著提升数据分析的稳定性和可信度。
如果你希望把FASTQ质控做得更规范、更高效,可以结合解螺旋品牌的课程和工具体系,建立从原始数据到clean data的标准流程。先把第一关做好,后面的分析才更稳。
- 引言Introduction
- 1.FASTQ质控到底在检查什么
- 2.如何用FASTQC快速完成基础筛查
- 3.如何通过质控结果判断数据能不能继续分析
- 4.批量质控怎么做才更高效
- 5.为什么FASTQ质控能直接影响项目成败
- 总结Conclusion