引言Introduction
VCF过滤是WES分析里最容易出错的一步。VCF里会混入低质量、污染、重复扩增和群体高频变异。若不处理好,后续注释和解读都会偏离临床与科研目标。掌握VCF过滤,决定你能否把真实突变从噪音里筛出来。 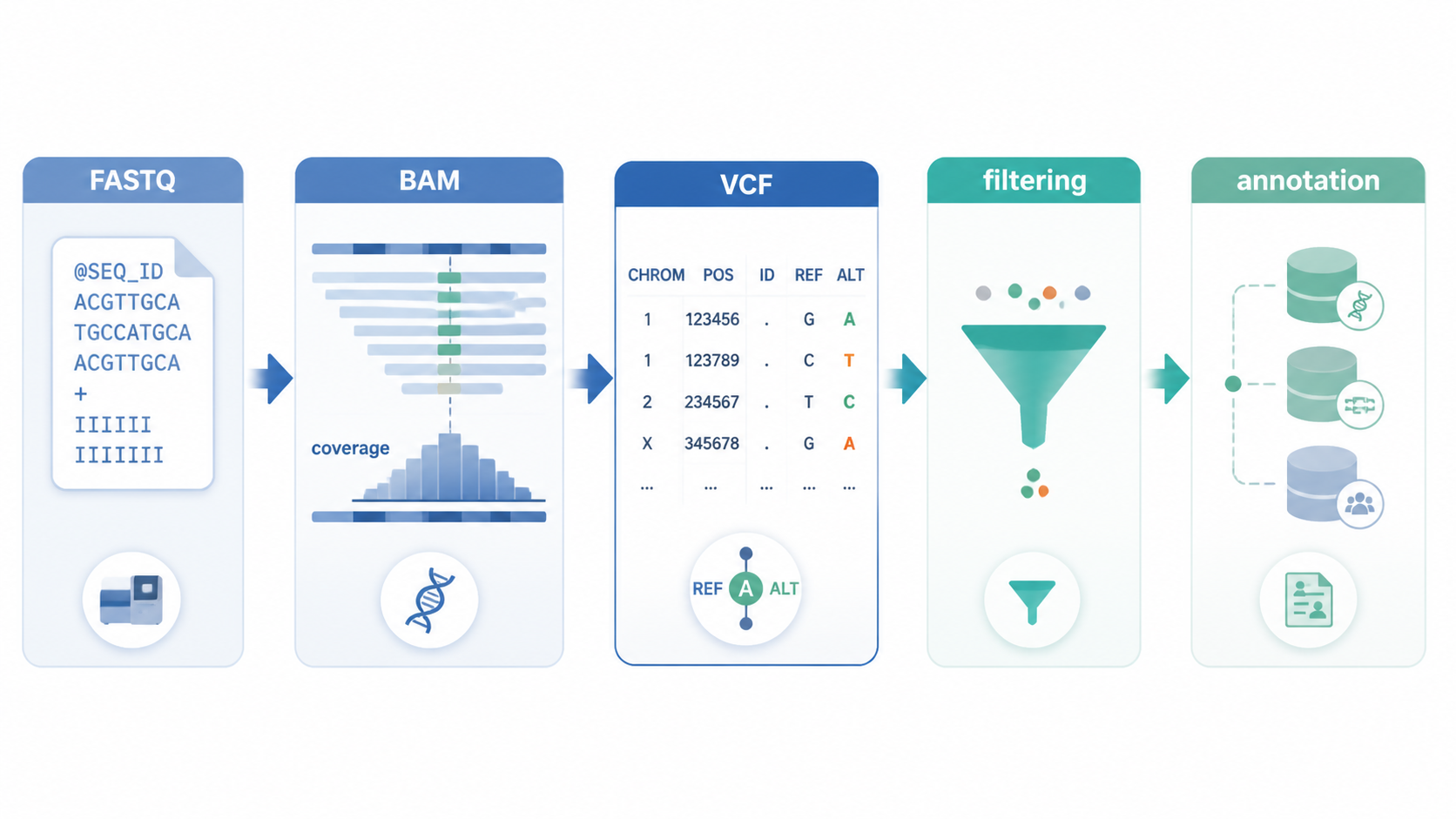
1.VCF过滤前,先确认输入数据质量
1.1 先看bam文件,再谈变异
VCF过滤不是孤立步骤。上游bam质量会直接影响最终结果。常看三个指标。
- coverage ,即平均覆盖度,反映外显子区域被reads覆盖的深度。
- mapping rate ,反映reads中有多少能比对到参考基因组。
- MAPQ ,反映reads的比对质量。
如果coverage不足、mapping rate偏低,后面的VCF过滤再严格,也只能得到“干净但不完整”的结果。
1.2 变异检测软件本身会影响过滤压力
不同variant calling软件的策略不同。GATK通常更严格,假阳性少,但可能漏掉部分真实变异。其他软件可能更“宽松”,召回更多候选位点,但噪音也更高。
因此,VCF过滤前要先知道:你的VCF是从什么调用流程来的。 这决定了后续阈值是否需要更保守或更宽松。
2.第一步:按VCF内置质量标记过滤
2.1 先保留PASS位点
VCF文件里通常会有质量标记。调用软件会根据内部规则给位点打标。最基础的做法是:
- 优先保留标记为PASS 的位点。
- 剔除明确被判定为低可信的记录。
- 对保留边界位点时,结合下游注释一起看。
这一步是VCF过滤的“第一道门”。它不解决全部问题,但能快速去掉明显不合格的候选。
2.2 关注clustered mutations
如果某一小段区域内,比如20 bp附近,突然聚集了很多变异,通常不正常。GATK会把这类情况标成clustered mutations 。
这类位点常提示局部测序误差、比对异常或文库问题。如果变异密度异常升高,应优先排查,而不是直接纳入解读。
3.第二步:排除比对质量差和边界异常位点
3.1 MAPQ低的位点要谨慎
MAPQ表示reads比对到参考基因组是否唯一、是否可靠。若reads存在多重比对,或者落在重复区域,MAPQ会下降。
在VCF过滤中,这类位点要重点关注,因为它们更容易出现假阳性。尤其是在复杂基因家族、重复序列和结构复杂区域,低MAPQ位点的解释价值明显下降。
3.2 read边界效应不能忽视
当变异位于read的两端边界附近时,更容易出现错误。即便测序质量看起来不差,边界位点仍可能受末端误差影响。
这也是为什么VCF过滤不能只看单一质量值。位点在read中的位置,本身就是一个重要风险信号。
4.第三步:去除污染与PCR假阳性
4.1 污染样本会制造“假变异”
污染可能来自样本之间交叉污染,也可能来自其他物种污染。对于肿瘤样本和混样样本,这个问题尤其要警惕。
如果污染存在,VCF里会出现不属于真实样本的变异信号。此时,简单提高阈值并不够,必须结合污染评估结果重新筛位点。
4.2 PCR重复引起的伪变异也要剔除
有些变异看似真实,实际上来源于PCR重复或扩增偏倚。尤其是局部重复序列、低复杂度区域,更容易出现这类问题。
在VCF过滤中,这类位点应优先排除。真实突变应该在独立reads中稳定出现,而不是被扩增过程放大。
5.第四步:结合群体频率过滤常见无害变异
5.1 高频人群变异通常优先过滤
VCF过滤离不开群体数据库。常见数据库包括千人基因组、gnomAD等群体频率资源。
如果某个位点在人群中频率较高,通常更可能是germline变异,或者对疾病没有明显贡献。
因此,分析肿瘤或遗传病相关数据时,高频位点通常应优先下调优先级或直接过滤。
5.2 阈值要随研究目的调整
不是所有高频位点都一定要删除。
- 做罕见病或肿瘤驱动突变分析时,通常更倾向过滤群体高频位点。
- 做药物反应或人群关联研究时,部分常见变异反而有意义。
所以,VCF过滤不是固定模板。阈值必须服务于研究问题。
6.第五步:区分肿瘤体细胞变异和胚系变异
6.1 配对样本模式更可靠
对于体细胞突变,最好使用肿瘤-正常配对模式。这样可以通过正常样本排除胚系变异。
如果某个位点在正常样本中也存在,就要高度怀疑它不是体细胞突变。
这是VCF过滤中最关键的生物学约束之一。
6.2 Tumor-only模式要更严格
如果没有正常对照,只能用Tumor-only模式。此时过滤压力更大,因为胚系变异更难去除。
通常需要结合:
- 群体频率数据库。
- Panel of Normals, PON。
- 位点深度与变异丰度。
- 已知肿瘤热点位点。
Tumor-only分析中,VCF过滤必须更谨慎,否则假阳性会明显升高。
7.第六步:用PON和数据库去除系统性伪位点
7.1 PON是去伪的重要工具
PON,panel of normals,指正常样本集合。它能帮助去除在正常样本里反复出现的系统性噪音位点。
这在SNV、CNV和SV分析中都很常见。
如果某个位点总是在正常样本中出现,就不应轻易当作真实突变。
7.2 数据库比对要做交叉验证
除了PON,还应参考ClinVar、ClinGen等临床数据库,以及项目相关的内部知识库。
对于致病性判断,数据库能帮助识别哪些变异更值得保留。
但要注意,数据库是证据,不是结论。 过滤时要结合样本类型、疾病背景和检测平台综合判断。
8.第七步:过滤后再做注释和人工复核
8.1 先过滤,再注释,效率更高
经过前面几轮VCF过滤后,保留下来的位点数量会明显下降。
这时再进行VEP、Annovar或SnpEff等注释,效率更高,结果也更清晰。
注释后可重点看:
- missense、frameshift、stop gain等功能类型。
- 致病性数据库证据。
- 与肿瘤或遗传病相关的基因。
8.2 关键位点必须回到原始比对图验证
再好的过滤规则,也不能替代人工复核。对临床相关位点,最好回看IGV。
重点检查:
- 是否有稳定的双链支持。
- 是否位于重复区。
- 是否存在明显偏倚。
- 是否与正常样本矛盾。
真正高质量的VCF过滤,最后一定要回到证据本身。
9.把VCF过滤做成可复用流程
9.1 建议的实操顺序
一个更稳妥的VCF过滤顺序通常是:
- 检查bam质量。
- 保留PASS位点。
- 过滤低MAPQ、边界异常、clustered mutations。
- 去除污染和PCR伪阳性。
- 按群体频率过滤。
- 用PON和正常样本排除系统性噪音。
- 结合注释和IGV复核。
这个顺序的核心原则是,先去技术噪音,再看生物学意义。
9.2 自动化比手工更适合大规模项目
WES报告和VCF分析常常涉及大量位点,人工逐条处理不现实。
对科研和临床项目来说,最好把VCF过滤写成标准化流程,统一参数、统一阈值、统一输出格式。
这样更利于重复分析、版本管理和结果追踪。
总结Conclusion
VCF过滤的本质,不是“删掉更多位点”,而是把技术噪音、系统伪影和常见无害变异尽可能分离出来 。真正有效的流程,通常包括7个关键步骤:先看bam质量,再筛PASS位点,处理比对异常,排除污染和PCR伪阳性,结合群体频率过滤,借助PON与正常样本去噪,最后再注释和人工复核。
如果你希望把这套流程做得更稳、更快、更适合临床或科研场景,可以借助解螺旋 的标准化分析与报告能力,把复杂的VCF过滤步骤串成可复用流程,减少漏检和误判,提升分析效率。

- 引言Introduction
- 1.VCF过滤前,先确认输入数据质量
- 2.第一步:按VCF内置质量标记过滤
- 3.第二步:排除比对质量差和边界异常位点
- 4.第三步:去除污染与PCR假阳性
- 5.第四步:结合群体频率过滤常见无害变异
- 6.第五步:区分肿瘤体细胞变异和胚系变异
- 7.第六步:用PON和数据库去除系统性伪位点
- 8.第七步:过滤后再做注释和人工复核
- 9.把VCF过滤做成可复用流程
- 总结Conclusion