引言Introduction

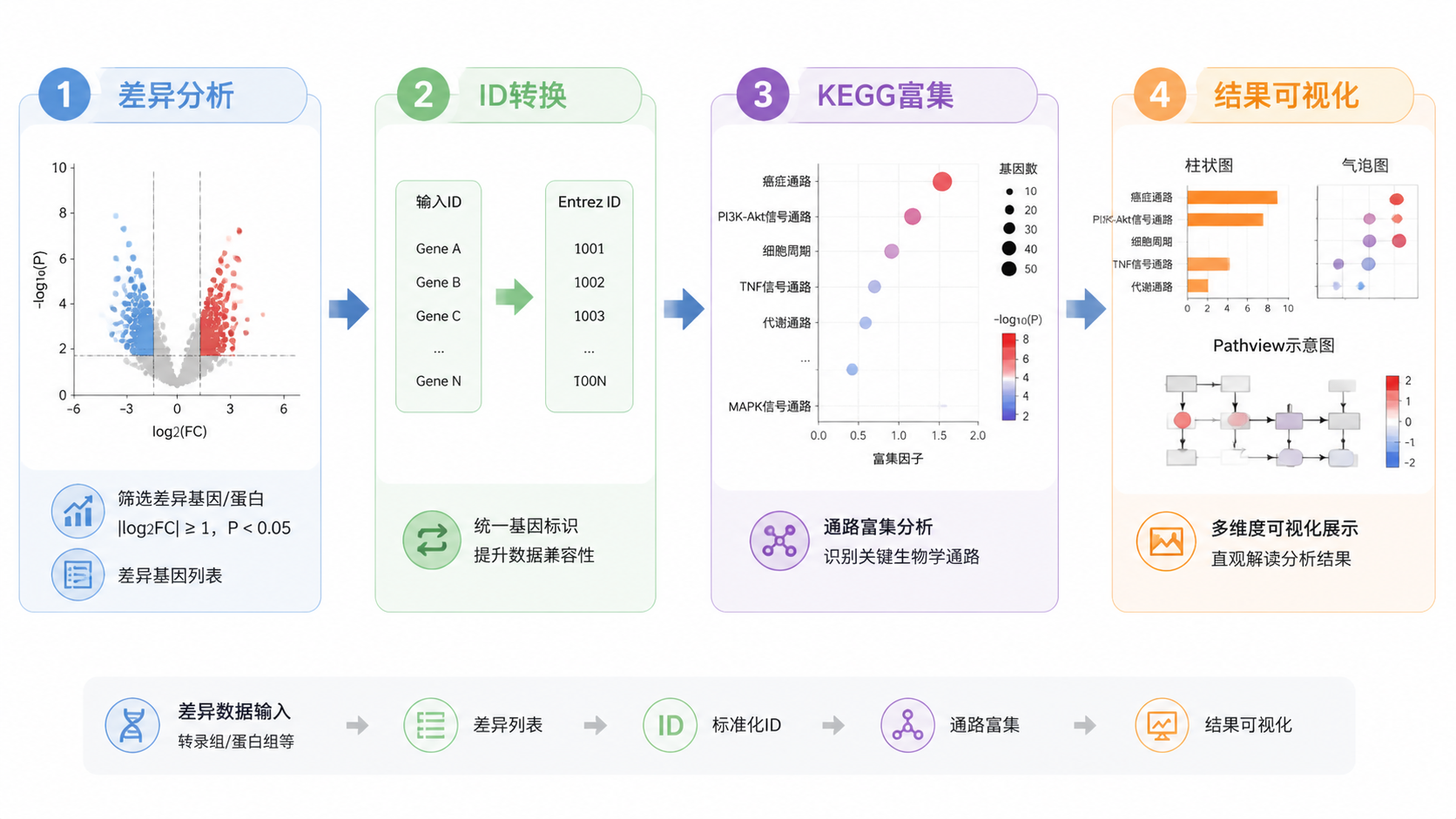
做完差异分析后,很多人卡在kegg富集分析。 基因列表有了,却不知道如何转换ID、如何解释通路结果、如何把图做得规范。本文用3步拆解核心逻辑,帮助医学生、医生和科研人员快速建立可复用的分析框架。
1. 先理解kegg富集分析到底在做什么
1.1 从“基因列表”走向“通路解释”
kegg富集分析的目标,不是简单罗列通路名,而是回答一个更关键的问题:这批差异基因是否集中影响了某些已知生物通路。
在实际研究中,常见输入是差异表达基因,输出则是显著富集的KEGG通路。它能帮助你把“基因变化”翻译成“生物学含义”。
以TCGA或单细胞下游分析为例,先得到上调或下调基因,再做富集。这样能更清楚地判断某条通路是被激活,还是被抑制。这一步决定了后续机制讨论是否站得住。
1.2 为什么ID转化是第一道门槛
很多人第一次做kegg富集分析时失败,不是因为算法,而是因为ID不对。
课程提纲里明确给出了转换路径:先把 ENSEMBL 转成 SYMBOL ,再转成 ENTREZID ,因为 enrichGO 和 enrichKEGG 这类函数通常依赖 Entrez ID。
常用代码思路如下:
genelist <- bitr(
up_genes,
fromType = 'SYMBOL',
toType = 'ENTREZID',
OrgDb = 'org.Hs.eg.db'
)
如果ID映射不完整,后面的富集结果就会缺失。 这会直接影响显著通路数量和可信度。
1.3 先做分组和差异分析,再谈富集
kegg富集分析不是独立步骤。它依赖前面的差异分析。
在课程示例中,先筛选肿瘤样本,再按基因表达分组,最后用 DESeq2 做差异分析。逻辑很清楚:先找出“变化的基因”,再判断这些基因“影响了什么功能”。
典型流程包括:
- 筛选样本。
- 提取表达矩阵。
- 设定分组。
- 建立 DESeq2 对象。
- 提取上调或下调基因。
- 进行通路富集。
没有可靠差异分析,kegg富集分析的解释价值会明显下降。
2. 按标准流程完成kegg富集分析
2.1 环境准备要先清干净
正式分析前,建议先清空环境变量,再加载必要R包。
课程中使用了 tidyverse、DESeq2、clusterProfiler、org.Hs.eg.db、enrichplot、ggplot2、pathview 等包。这个组合已经覆盖了差异分析、ID转换、富集分析和可视化。
rm(list = ls())
library(tidyverse)
library(DESeq2)
library(clusterProfiler)
library(org.Hs.eg.db)
library(enrichplot)
library(ggplot2)
library(pathview)
环境统一,结果才更容易复现。 对科研写作和组内协作尤其重要。
2.2 富集分析核心参数要看懂
在 kegg富集分析 中,最关键的是输入基因、物种和显著性阈值。
课程中给出的写法是:
kk <- enrichKEGG(
gene = genelist$ENTREZID,
organism = 'hsa',
pAdjustMethod = 'BH',
pvalueCutoff = 0.05,
qvalueCutoff = 0.25
) %>%
setReadable(OrgDb = org.Hs.eg.db, keyType = 'ENTREZID')
这里有几个要点:
- gene 是输入基因列表。
- organism = ‘hsa’ 代表人类。
- pAdjustMethod = ‘BH’ 是多重检验校正。
- setReadable 可把结果转回更易读的基因名。
如果同时做GO和KEGG,可把两个结果合并,便于后续统一展示。课程里使用了 merge_result() 和 replace_na() 来整理结果表。这一步是为了让BP、CC、MF和KEGG在同一张结果表中呈现。
2.3 先取Top条目,再做图,信息更集中
很多初学者一上来就展示全部结果,图会很乱。
更好的做法是先按类别筛选 top 5 或 top 10,再可视化。课程中按 BP、CC、MF、KEGG 分别提取前 5 条,再合并成 category_gokegg。
这样做有两个好处:
- 结果更聚焦。
- 图更适合论文和汇报。
对于kegg富集分析来说,Top条目不是“全部答案”,但足够用来判断主要生物学方向。
3. 学会用图读懂kegg富集分析结果
3.1 柱状图和气泡图分别回答什么问题
课程里给出了两种常用图:柱状图和气泡图。
柱状图更适合看 GeneRatio 的排序。气泡图则同时展示 GeneRatio、显著性和基因数,信息更丰富。
柱状图代码思路如下:
ggplot(category_gokegg, aes(
x = factor(Description, levels = rev(unique(category_gokegg$Description))),
y = unlist(lapply(GeneRatio, function(x) { eval(parse(text = x)) })),
fill = p.adjust
)) +
geom_bar(stat = 'identity') +
coord_flip()
气泡图则常用:
ggplot(category_gokegg, aes(
x = factor(Description, levels = rev(unique(category_gokegg$Description))),
y = unlist(lapply(GeneRatio, function(x) { eval(parse(text = x)) })),
color = p.adjust,
size = Count
)) +
geom_point(stat = 'identity') +
coord_flip()
读图时优先看 p.adjust、Count 和 GeneRatio。 这三项比单纯看通路名字更重要。
3.2 网络图和热图能补足什么信息
如果想进一步看“哪些基因驱动了通路”,可以用网络图和热图。
课程中使用 cnetplot() 和 heatplot() 来展示通路与基因的对应关系。前者适合看基因与通路的连接结构,后者适合看不同通路中基因的表达变化。
例如:
- 网络图可以帮助你找出一个通路是否由少数关键基因主导。
- 热图可以帮助你判断上调与下调方向是否一致。
这对机制分析很重要。 尤其在肿瘤、免疫和单细胞研究中,通路不是终点,关键是找到可解释的基因集合。
3.3 Pathview适合做通路落图
如果你需要把差异基因映射到具体 KEGG 通路图上,pathview() 很实用。
课程里通过 pathwayID 逐个循环绘制通路图,便于查看基因在代谢或信号通路中的位置变化。
pv.out <- pathview(
gene.data = -log10(up_padj),
pathway.id = i,
species = 'hsa',
out.suffix = 'pathview'
)
这类图适合论文补充材料,也适合在组会中展示。它能把抽象的富集结果变成直观的通路变化图。
4. 真正做好kegg富集分析,关键是这3个判断
4.1 判断一:输入基因是否足够可靠
富集结果强不强,首先取决于输入基因。
如果差异阈值设得太松,假阳性会增加。若阈值太严,可能漏掉真实信号。课程中提到的常见标准是按 p 值和 logFC 筛选显著基因,再做富集。输入质量决定输出质量。
4.2 判断二:物种和ID是否一致
kegg富集分析里,物种参数很关键。
人类数据常用 hsa。如果物种选错,结果会直接偏离。ID类型也必须统一,尤其是在 ENSEMBL、SYMBOL、ENTREZID 之间转换时,要检查重复和缺失。
课程中还特别处理了重复项:
genelist <- genelist[!duplicated(genelist$ENSEMBL), ]
这能减少重复映射造成的偏差。
4.3 判断三:结果是否能回到生物学问题
富集分析不是终点。
你需要回到原始课题,比如肿瘤增殖、免疫浸润、代谢重编程或信号通路异常。课程示例中,通路结果会帮助研究者判断后续方向。比如某些免疫相关通路富集明显,就提示可以继续做免疫机制验证。
真正有价值的kegg富集分析,一定能服务于下一步实验设计。
总结Conclusion
kegg富集分析的核心逻辑很简单,只有3步:先准备可靠的差异基因,再完成标准化的ID转化,最后用合适的图形解释通路结果。
对医学生、医生和科研人员来说,它的价值不只是“出图”,而是把分子数据转成可讨论的机制证据。只要把输入、参数和可视化三件事做好,分析就会更稳、更清晰,也更容易写进论文和课题汇报。
如果你希望把 kegg富集分析 做得更快、更规范,可以直接使用解螺旋的课程与实操资源。 它能帮助你少走弯路,快速搭建从差异分析到通路解读的完整流程。
- 引言Introduction
- 1. 先理解kegg富集分析到底在做什么
- 2. 按标准流程完成kegg富集分析
- 3. 学会用图读懂kegg富集分析结果
- 4. 真正做好kegg富集分析,关键是这3个判断
- 总结Conclusion