引言Introduction
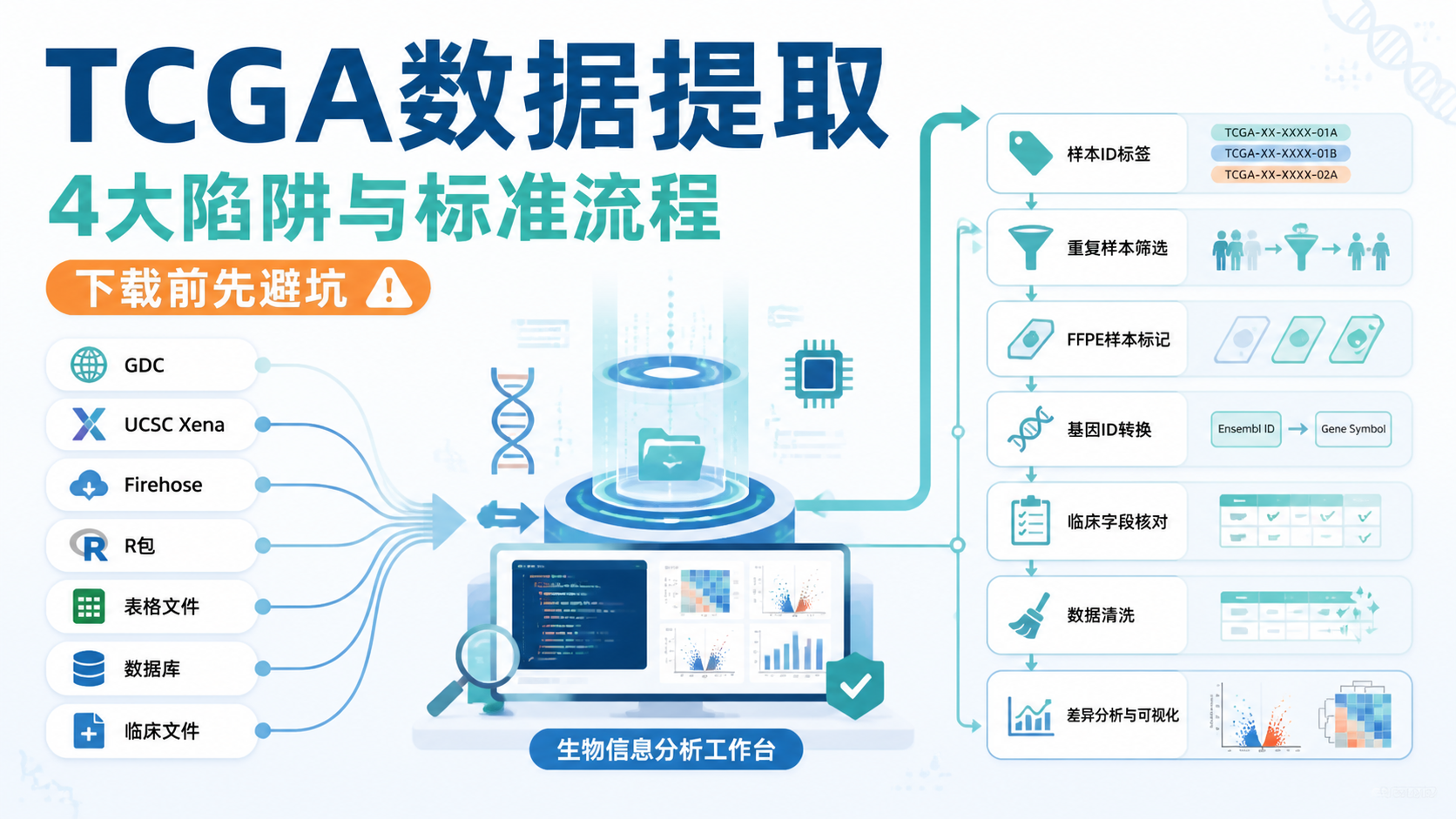
TCGA数据格式转换为什么总出错?很多人不是不会分析,而是卡在分期、样本ID、文件类型和重复样本处理上。同一批TCGA数据,XML、File Tab、barcode、legacy、harmonized、FPKM、counts,任何一步理解错,后面都会连锁报错。
1. TCGA数据格式转换最常见的报错来源
1.1 文件类型选错,临床信息就会变复杂
在TCGA里,临床数据并不只有一种来源。常见有XML和File Tab两类。问题在于,XML里分期字段往往更细,容易出现stage 3、3A、3B、3C这类层级。对于tcga数据格式转换 来说,这类字段一旦直接切分字符串,很容易把临床分期解析错。
相比之下,TCGA官方更推荐File Tab格式。它把临床、随访、放疗、用药等信息拆分得更清楚。如果你的目标是提取病理分期,优先使用整理好的clinic patient文件,会比XML解析稳定得多。
1.2 clinical stage和pathological stage混用
这是最容易出错的地方之一。临床分期是医生根据临床表现判断的分期,病理分期是基于病理标本确认的分期。两者不是一回事。课程知识库明确指出,我们在分析中需要关注的是pathological stage,而不是clinical stage。
例如,TNM里常见的目标信息包括T3、N x、M0。若把clinical stage误当作pathological stage,最终纳入的数据就会偏离真实病理结果。做tcga数据格式转换时,先确认分期来源,再做下游整理。
1.3 stage字段层级太细,直接拆分容易丢信息
TCGA临床字段里,stage常常不是单一值,而是带细分类别。比如stage 3下面会细分为3A、3B、3C。N分期也可能出现更细的子类。若你只是按“stage 3”粗暴截取,可能会漏掉子分类信息,或者误归并不同患者。
更稳妥的做法是先明确研究目标,再决定保留层级。如果研究只关注病理三期,就统一提取pathological stage的stage 3。 如果要做更细分层分析,建议保留原始字段,不要提前压缩。
2. TCGA数据格式转换中,barcode和样本重复最容易引发混乱
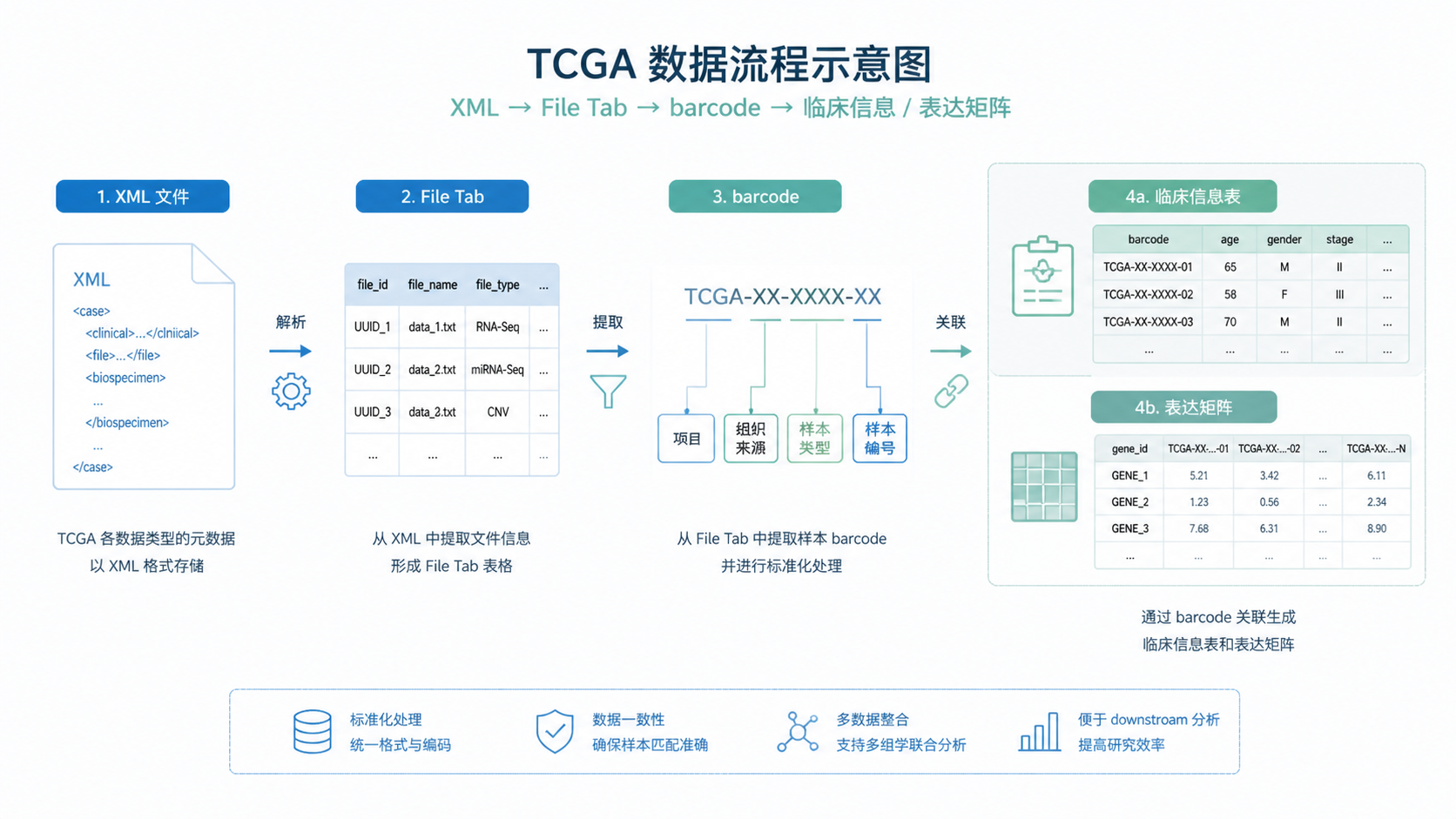
2.1 barcode不是简单编号,而是样本身份规则
TCGA的barcode是样本识别核心。它不仅表示项目来源,还包含组织来源、样本类型、分析类型、板号等信息。很多格式转换错误,本质上是没有读懂barcode。
其中最关键的是sample字段。它用数字区分样本类型。知识库提到,tumor样本通常是01到09,normal是10到19,control是20到29。也就是说,以0开头的样本通常属于tumor。 如果你在格式转换时没有正确拆解barcode,后续做肿瘤和正常配对就会错位。
2.2 同一个病人出现多个样本,不能直接全保留
TCGA经常存在重复测序或重复样本问题。表面上看,barcode前四位甚至更长部分相同,但plate、center或analyte不同。这个时候不能简单合并,更不能直接全部保留。
课程知识库给出的原则是,要结合analyte replicate filter和sort replicate filter来筛选。先按样本类型过滤,再按测序批次和板号判断保留哪个版本。 如果两个样本实际上是同一病人同一组织的重复测序,通常保留后者,更符合后续整理规则。
2.3 FFPE样本和重复板号也会制造假冲突
部分样本还会因为FFPE处理、不同plate或不同center而产生重复记录。此时如果不做清洗,临床合并时就会出现一对多、一对零的问题。tcga数据格式转换失败,很多时候不是数据坏了,而是重复样本没有先处理。
建议在合并前先做三步检查。
- 提取样本前16位或更稳定的唯一标识。
- 检查是否存在重复barcode。
- 按官方规则筛除重复和低质量样本。
3. 表达数据格式转换出错,常见于counts、FPKM和LOG2处理
3.1 不同数据层级不能混用
TCGA数据里,counts、FPKM、FPKM-UQ、normalized数据并不是同一层级。counts更适合DESeq2和edgeR这类差异分析,FPKM类数据更偏表达量展示和部分下游分析。 如果把已经标准化的数据当作原始counts用,分析结果会偏。
知识库中提到,UCSC Xena网站的数据下载较方便,而且与GDC整理后的CONS数据一致。对于需要整数矩阵的分析,先确认输入是否已经经过LOG2处理很重要。如果是LOG2后的数据,必须先逆LOG再进行整数化处理。
3.2 读入格式不一致,会导致整列错位
TCGA下载文件常见TSV和CSV两种格式。实际处理中,文件分隔符不统一是高频报错原因。读取时先尝试TSV,失败再试CSV。如果都失败,就要打开原文件检查分隔方式。
很多“格式转换报错”,本质是分隔符、列名、行名三者不一致。 比如行名是gene ID,列名是样本barcode,若读取函数没有正确设置,会把第一列当数据,导致矩阵整体偏移。
3.3 ID转换后基因数变化是正常现象
知识库提到,example ID转换前后,基因数会从6万多变成2万多。这并不一定是错误,而是ID映射和过滤后的正常结果。不同版本的注释、不同转录本来源、不同基因模型,都会影响最终保留的基因数。
所以做tcga数据格式转换 时,不能只看“数量变少”就判断失败。要先核对映射规则,再看是否保留了目标基因集。
4. legacy和harmonized版本不同,版本差异也会导致转换失败
4.1 参考基因组版本不同,坐标就不一样
TCGA有legacy数据和GDC harmonized数据之分。两者在参考基因组上存在差异。知识库指出,legacy常见于HG19/GRC37,而harmonized更多对应HG38/GRC38。如果你把两个版本的数据直接拼接,坐标和注释很容易不一致。
这类问题常出现在拷贝数、甲基化、突变注释和转录组比对中。即使同一项目,不同版本的数据仍可能因坐标转换和质控策略不同而产生差异。
4.2 新旧流程差异会影响结果一致性
GDC整理后的数据采用了更新的流程。比如RNA-seq重新使用STAR比对,突变调用用Mutect2替代旧流程中的Mutect,基因注释也进行了升级。这意味着旧版和新版不是简单换个文件名,而是整条处理链不同。
好消息是,知识库也提到,参考基因组更换后,结果仍然具有较高一致性。但前提是,你要在同一版本体系内完成分析。不要把legacy和harmonized数据混合成一个矩阵。
4.3 官网数据通常更新更及时
TCGA官网和GDC数据更新更快,临床bug修复也更及时。知识库明确建议,优先从官网下载最新数据,或者使用UCSC Xena整理好的数据。如果你的tcga数据格式转换经常出错,先检查数据源是否过旧。
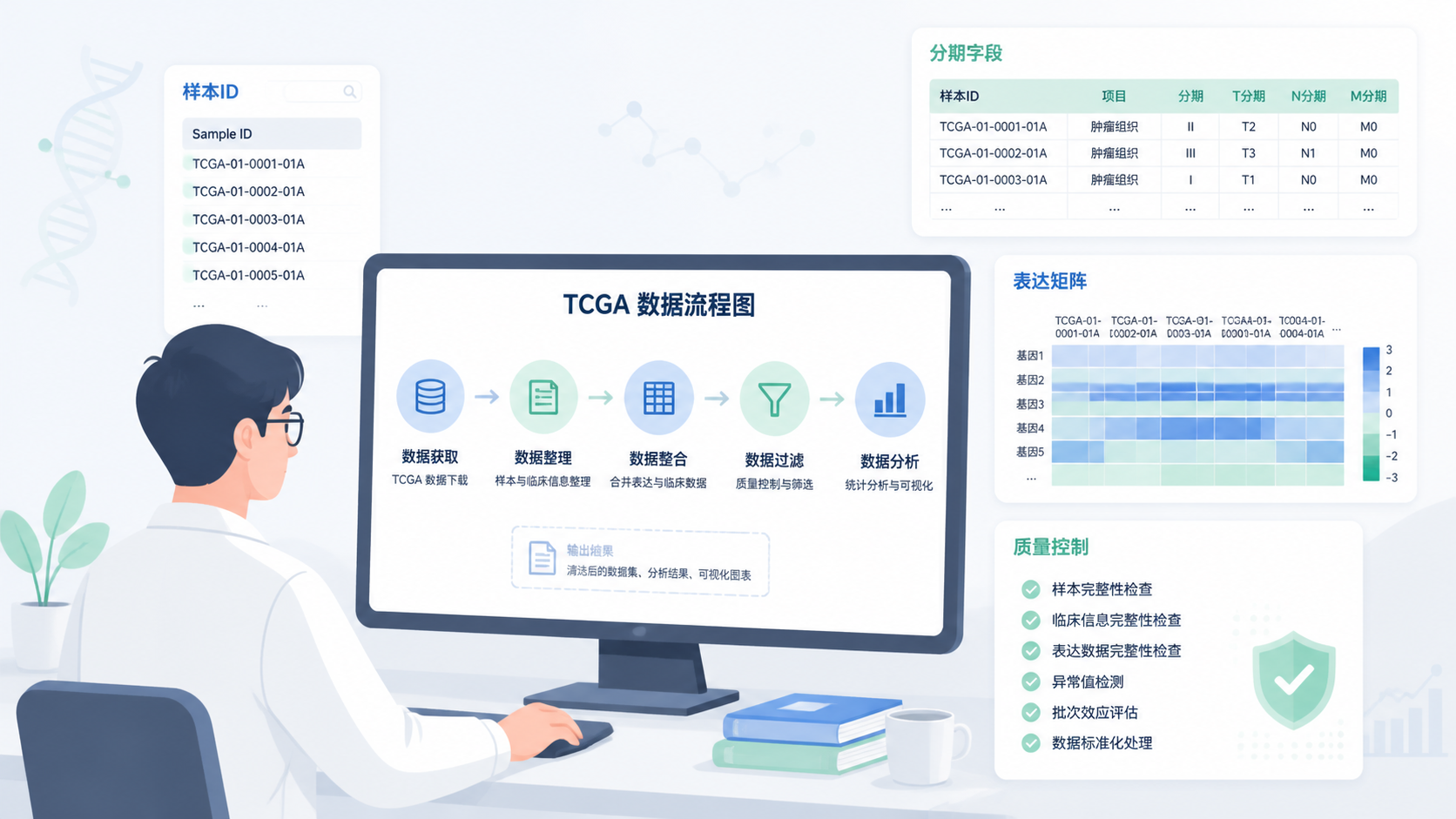
5. 一套更稳妥的TCGA格式转换流程
5.1 先定分析目标,再选字段
做任何转换前,先回答三个问题。
- 你要的是临床信息,还是表达矩阵。
- 你要的是clinical stage,还是pathological stage。
- 你要的是counts,还是FPKM,还是生存信息。
目标不清,字段就会选错。 这一步能直接减少后面一半错误。
5.2 统一样本ID,再做临床合并
样本ID是TCGA合并的核心。建议先规范到统一长度,再提取病人层级和样本层级信息。对于重复测序样本,优先按官方规则筛选,再与临床表合并。
可执行步骤如下。
- 先检查barcode是否重复。
- 再筛除不需要的样本类型。
- 然后按病人ID合并临床信息。
- 最后再做表达矩阵与生存信息整合。
5.3 数据过滤要放在合并前
知识库强调,样本过滤和基因过滤都很重要。样本层面要剔除不合格或不需要的条目。基因层面通常过滤低表达基因,以提高差异分析敏感性和准确度。
不要先大规模合并,再回头清洗。 那样一旦发现分期、barcode或重复样本错误,整个流程都要重做。
总结Conclusion
TCGA数据格式转换之所以总出错,核心原因不是工具不够多,而是临床分期、barcode规则、样本重复、数据版本和表达矩阵层级 没有先理清。对医学生、医生和科研人员来说,最稳妥的策略是先确认pathological stage,再统一样本ID,再区分legacy和harmonized版本,最后选择合适的表达数据类型。
如果你希望把这些步骤做得更快、更稳,建议使用解螺旋品牌的生信内容与数据整理支持方案,减少临床字段误读、样本ID错配和格式转换返工。把复杂的TCGA数据流程交给更清晰的方法,才能把时间留给真正的科研分析。
- 引言Introduction
- 1. TCGA数据格式转换最常见的报错来源
- 2. TCGA数据格式转换中,barcode和样本重复最容易引发混乱
- 3. 表达数据格式转换出错,常见于counts、FPKM和LOG2处理
- 4. legacy和harmonized版本不同,版本差异也会导致转换失败
- 5. 一套更稳妥的TCGA格式转换流程
- 总结Conclusion