引言Introduction
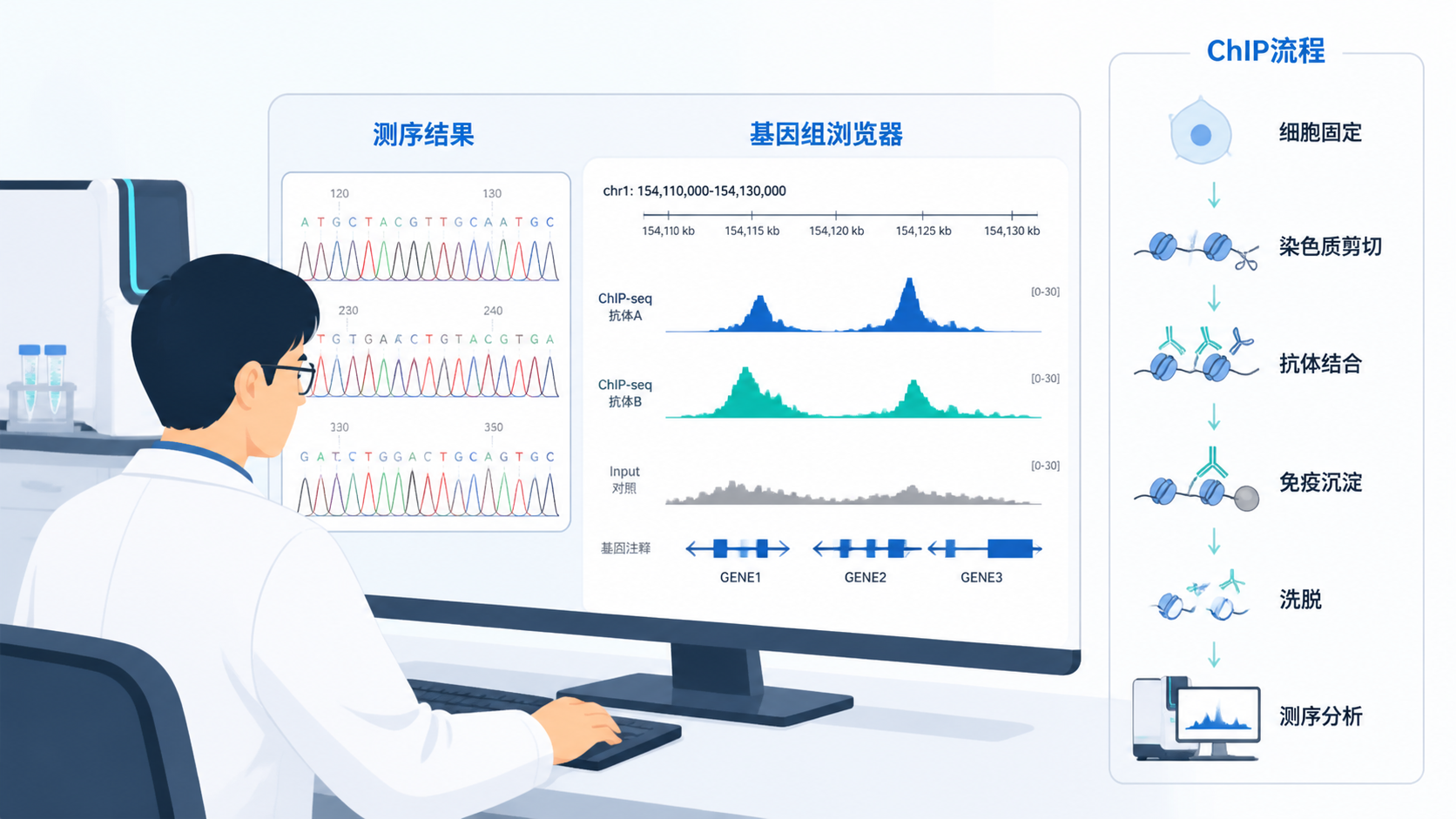
Chip-seq峰值数据 是转录因子结合位点、组蛋白修饰分布分析的核心输出。很多人做完实验后,真正卡住的不是测序,而是峰值是否可靠、是否可复现、是否能用于后续机制研究。本文从实验设计、数据分析到结果质控,系统说明如何获得高质量Chip-seq峰值数据 。
1. 理解Chip-seq峰值数据的来源
1.1 峰值数据本质上来自富集信号
Chip-seq的产物是纯化的DNA。它不是直接给出“峰”,而是先得到测序片段,再通过比对和峰值识别算法,推断蛋白结合区域。也就是说,Chip-seq峰值数据 是分析流程的结果,不是实验结束后的自然产物。
峰值质量,首先取决于免疫沉淀是否真正富集到了目标区域。 如果抗体特异性差,或染色质片段化不均匀,后续再好的软件也难以挽救结果。
1.2 不同实验类型对应不同输出形式
ChIP-seq、ChIP-PCR、ChIP-qPCR的数据形态并不相同。
- ChIP-seq原始数据是测序数据,需要经过专业软件和数据库分析。
- ChIP-PCR原始数据常表现为电泳后的条带图。
- ChIP-qPCR原始数据与普通qPCR一致,可进一步获得Ct值并绘制柱状图。
因此,讨论Chip-seq峰值数据 时,重点在于测序、比对、峰值识别和质控的完整链路,而不是只看最终峰图。
2. 高质量峰值数据的前提:实验设计要正确
2.1 选择合适的抗体和对照
抗体质量是决定峰值可信度的第一道门槛。 优先选择文献验证充分、ChIP级别明确的抗体。若抗体不适配ChIP应用,常见问题包括背景高、峰少、重复性差。
对照也必须规范。通常需要输入对照(Input),用于评估背景富集。没有合格对照,峰值容易出现假阳性,影响Chip-seq峰值数据 的解释。
2.2 控制样本量、重复和生物学一致性
高质量数据不是“测一次就够”。至少应设置生物学重复。重复之间相关性越高,峰值越稳定。若重复差异过大,说明实验流程存在系统误差。
在样本准备阶段,还要尽量统一以下条件:
- 细胞状态一致。
- 交联条件一致。
- 超声片段长度相近。
- 免疫沉淀和洗脱条件稳定。
这些细节会直接影响Chip-seq峰值数据 的峰高、峰宽和背景噪声。
3. 影响峰值质量的关键实验环节
3.1 染色质片段化要均一
片段化不均会导致峰值偏移,甚至影响峰形。理想情况下,染色质应被打断到适合建库的范围,并保持分布相对集中。过长会降低分辨率,过短则可能损失真实信号。
均一片段化是获得清晰峰形的基础。 这一步做不好,后续峰值边界会变得模糊,尤其不利于转录因子这类窄峰分析。
3.2 免疫沉淀过程要减少非特异结合
洗涤不充分会增加背景,洗涤过强又可能丢失真实结合片段。需要在信号保留和背景控制之间找到平衡。对低丰度靶标尤其如此。
如果实验中背景条带明显、测序后比对率不稳定,Chip-seq峰值数据 往往会表现为“峰很多,但不可信”。
3.3 建库质量决定最终可分析性
建库中接头连接、片段筛选、PCR扩增都可能引入偏倚。过度扩增会造成重复率过高,降低峰值分辨率。文库复杂度不足时,即使测序深度增加,真实信息增量也有限。
高质量峰值数据要求文库复杂度足够高,重复率足够低。
4. 数据分析如何影响峰值结果
4.1 原始数据必须先完成基础质控
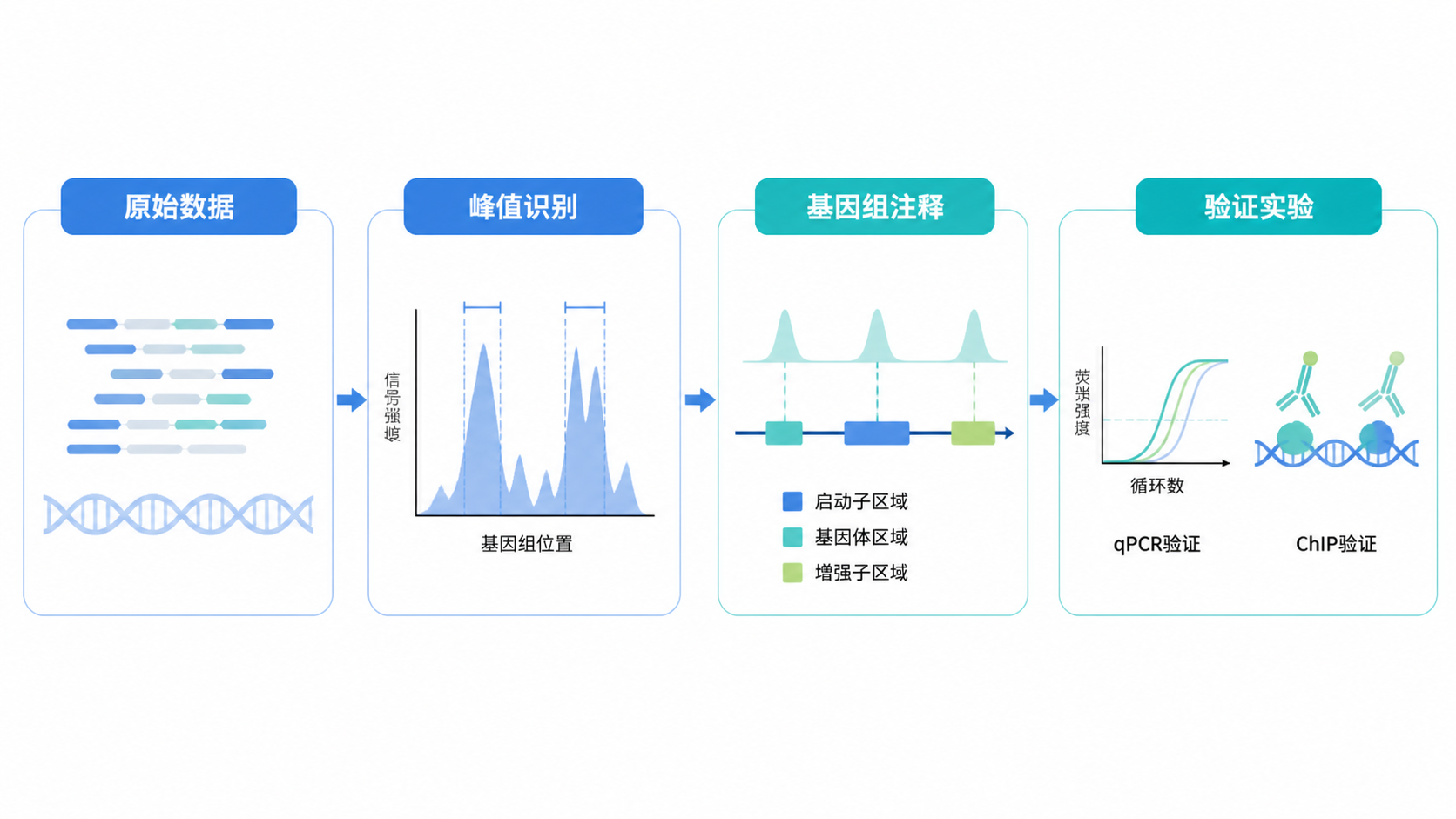
ChIP-seq原始数据通常是FASTQ文件,先要做reads质量评估、接头去除和低质量过滤。之后再比对到参考基因组。基础质控不过关,峰值识别结果会被噪声污染。
分析时应重点关注:
- 测序质量分布。
- 比对率。
- 唯一比对率。
- 重复率。
- 片段长度分布。
这些指标共同决定Chip-seq峰值数据 是否具有继续分析的价值。
4.2 峰值识别要匹配实验类型
转录因子通常形成窄峰,组蛋白修饰常出现宽峰。不同靶标需要选用合适的peak calling策略。若算法不匹配,可能把真实结合位点切碎,或把背景误判为峰。
算法选择不是技术细节,而是结果正确性的核心。
常见分析思路包括:
- 比较ChIP样本与Input的富集差异。
- 识别显著富集区。
- 合并重复并过滤低可信峰。
- 注释峰位于启动子、增强子还是基因体区域。
4.3 结果可视化要与统计结论一致
峰图、热图、metagene图和基因组浏览器截图,都是展示Chip-seq峰值数据 的重要方式。但可视化必须建立在统计显著性之上,不能只凭“看起来有峰”。
如果峰图漂亮,但重复间一致性低,或富集倍数不足,那么结果仍不适合直接用于机制发表。
5. 如何判断峰值数据是否高质量
5.1 看重复一致性
重复样本之间相关性高,说明实验稳定。相关性低,则可能存在样本污染、免疫沉淀效率波动或测序深度不足的问题。
高质量 的Chip-seq峰值数据 ,应该在重复间呈现稳定的峰位和相近的富集模式。
5.2 看背景噪声和峰形
好数据往往具备较低背景和清晰峰形。窄峰应集中,宽峰应连续。若出现大量散点式峰值,常提示背景过高或阈值设置过宽。
5.3 看后续生物学解释是否合理
真正有价值的峰值,应能支持后续问题。例如:
- 是否富集在启动子附近。
- 是否与已知调控基因相关。
- 是否和表达变化一致。
- 是否支持某条信号通路或转录调控模型。
如果峰值与生物学现象完全脱节,即便数量很多,也不算高质量Chip-seq峰值数据 。
6. 面向科研发表的实用建议
6.1 从一开始就按发表标准设计
高水平文章需要的不只是“做出来”,而是“可解释、可复现、可审查”。建议在实验前就明确:
- 靶标类型。
- 对照设置。
- 重复数量。
- 预期峰型。
- 后续验证方式。
发表级别的数据,必须把实验和分析当作一个整体。
6.2 用验证实验增强可信度
ChIP-seq发现峰值后,建议用ChIP-qPCR对关键位点做验证。因为ChIP-qPCR可以提供Ct值层面的定量证据,帮助确认富集是否真实。必要时还可结合电泳条带和柱状图展示。
这类多层验证能显著提升Chip-seq峰值数据 的可信度,也更符合E-E-A-T对专业性和信任度的要求。
6.3 借助规范化工具提升分析效率
对于缺少生信经验的科研人员,选择稳定、标准化的分析工具很重要。规范流程可以减少手工误差,也能让峰值识别、注释和可视化更统一。
如果希望更高效地整理Chip-seq峰值数据 ,可以借助解螺旋的相关产品或服务,把原始数据处理、结果呈现和后续验证串联起来,减少重复劳动,把精力集中在机制解读上。
总结Conclusion
高质量Chip-seq峰值数据 ,不是单靠测序深度堆出来的,而是由抗体、对照、片段化、建库、质控和峰值算法共同决定。想要结果可靠,必须从实验设计开始控制变量,再用规范的数据分析和必要的验证实验闭环确认。
如果你正在为峰值不稳定、背景过高、重复性差而困扰,解螺旋可以帮助你把ChIP-seq流程做得更规范、更高效。 从原始数据整理到结果展示,再到后续验证,都能更有条理地推进。
- 引言Introduction
- 1. 理解Chip-seq峰值数据的来源
- 2. 高质量峰值数据的前提:实验设计要正确
- 3. 影响峰值质量的关键实验环节
- 4. 数据分析如何影响峰值结果
- 5. 如何判断峰值数据是否高质量
- 6. 面向科研发表的实用建议
- 总结Conclusion