






引言Introduction
GFF注释信息是基因组研究中最常见、也最容易被忽视的数据之一。很多医学生、医生和科研人员拿到文件后,面对坐标、层级关系和属性字段,往往不知道从哪里下手。如果不能快速解析GFF注释信息,就很难把基因位置、外显子结构和功能注释真正用起来。 本文将用清晰步骤说明如何读取、理解并应用GFF注释信息。
1. 先理解GFF注释信息的核心结构
1.1 GFF文件到底记录了什么
GFF,全称General Feature Format,常用于描述基因组上的功能特征。它不是纯序列文件,而是把“特征”和“坐标”对应起来。常见内容包括基因、转录本、外显子、CDS、UTR等。
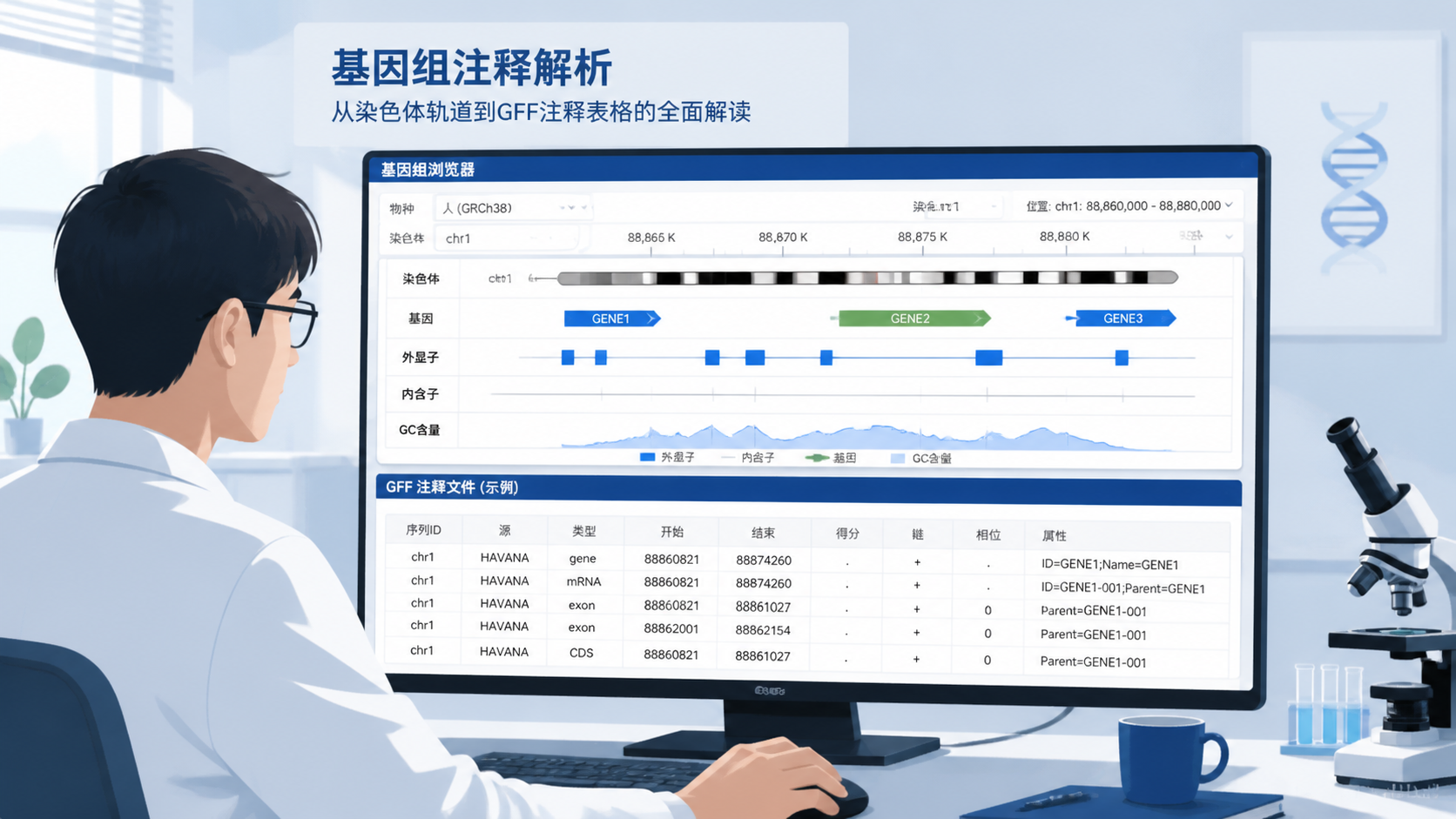
一条标准记录通常包含9列:序列名、来源、特征类型、起始位置、终止位置、得分、链方向、相位和属性。其中最关键的是第3、4、5和9列。 前者决定“是什么”,后者决定“在哪里、属于谁”。
1.2 为什么GFF注释信息适合基因组分析
GFF注释信息的价值在于,它把离散的序列片段组织成层级结构。比如一个gene下面可以有多个transcript,每个transcript又包含多个exon和CDS。这样的组织方式,特别适合转录组分析、变异注释和功能基因定位。
对临床科研来说,这种结构有实际意义。你可以快速判断一个变异是否落在编码区,是否影响剪接位点,是否位于已知功能域附近。这也是GFF注释信息比单纯FASTA更适合下游分析的原因。
2. 快速读懂GFF注释信息的9列
2.1 第1到第5列:位置和类型信息
第1列通常是染色体或contig名称。第3列是feature类型,常见值有gene、mRNA、exon、CDS。第4和第5列给出坐标范围,注意GFF通常采用1-based闭区间坐标。
这意味着起始和终止位点都包含在内。解析时如果按0-based思维处理,就容易出现偏移错误。这是新手最常见的坑之一。
2.2 第6到第8列:得分、链方向和相位
第6列是得分,很多注释文件中会留空或用“.”表示未知。第7列表示链方向,正链为“+”,负链为“-”。第8列相位主要用于CDS,帮助确定密码子阅读框。
如果你在做蛋白编码区分析,第8列不能忽略。它决定了CDS拼接后是否保持正确阅读框。对于变异效应预测而言,这一列会直接影响解释结果。
2.3 第9列:属性字段最关键
第9列是属性字段,通常以分号分隔,里面包含ID、Parent、Name、gene_id、transcript_id等信息。它决定不同条目之间的隶属关系。
例如,gene的ID可以作为上层节点,mRNA通过Parent字段指向该gene,exon再通过Parent指向转录本。只有把这层关系理顺,GFF注释信息才真正可用于可视化和统计分析。
3. 如何快速解析GFF注释信息
3.1 先做格式检查
解析前先确认文件是否为标准GFF3或GTF。二者结构相似,但属性字段写法不同。GFF3更强调ID和Parent,GTF则常见gene_id和transcript_id。
建议先检查三件事。
- 是否为制表符分隔。
- 是否每行都完整保留9列。
- 是否存在注释版本冲突或坐标异常。
格式不规范,后续所有统计都可能出错。
3.2 用常见工具完成快速筛查
对大多数科研场景,命令行工具是最高效的。可以先用grep、awk、cut做初步统计,再用gffread、AGAT或bedtools进行标准化和转换。若是可视化需求,可导入IGV、JBrowse或UCSC Genome Browser。
一个实用流程是:
- 先统计feature类型分布。
- 再检查gene和transcript的层级关系。
- 最后验证坐标与参考基因组是否匹配。
这样能在最短时间内发现文件缺失、重复或错配问题。
3.3 重点检查注释一致性
解析GFF注释信息时,最常见的问题不是“打不开”,而是“能打开但不一致”。例如一个exon没有Parent,或者CDS坐标超出转录本范围。还有一种情况是同一基因的多个转录本命名规则不统一。
这类问题会导致下游软件报错,或者产生错误统计。因此,真正的快速解析,不是只看内容,而是验证结构是否自洽。
4. GFF注释信息的典型应用场景
4.1 变异注释与功能判断
在医学和转化研究中,GFF注释信息最常用于判断突变落点。你可以把VCF与GFF结合,识别变异位于外显子、内含子、UTR还是启动子附近。对于编码区变异,还能进一步判断是否改变氨基酸。
这一步对于筛选候选致病位点非常重要。尤其在遗传病、肿瘤突变和药物基因组学研究中,GFF注释信息是基础入口。
4.2 转录本结构分析
如果研究关注可变剪接,GFF注释信息能帮助你比较不同转录本的外显子组成。你可以计算外显子数量、CDS长度、UTR长度,并比较不同isoform之间的结构差异。
这对研究组织特异性表达、疾病相关剪接事件非常有价值。很多论文中的“结构图”本质上都来源于GFF注释信息的二次整理。
4.3 基因组可视化与报告撰写
在写文章或汇报时,单纯列坐标没有说服力。把GFF注释信息导入基因组浏览器后,你可以直观看到基因结构、变异位置和功能区域。图形化结果更容易被同行和临床团队理解。
对于需要发表论文或制作基金申请材料的科研人员,这一步能显著提升结果表达质量。
5. 提高解析效率的实用方法
5.1 建立统一的标准流程
建议把GFF注释信息处理固定成一套流程。
- 下载原始注释文件。
- 核对版本号和参考基因组。
- 检查格式与层级。
- 转换为下游分析所需格式。
- 做可视化和抽样验证。
这样的流程适合课题组复用,也能减少不同成员之间的结果偏差。标准化,往往比单次手工处理更重要。
5.2 用脚本做批量提取
如果你经常处理多个样本或多个物种,建议用Python、R或Shell脚本做批量提取。比如按gene类型筛选、提取特定染色体上的注释、统计每个基因的外显子数,都是高频任务。
脚本化处理的好处很直接:
- 速度更快。
- 错误更少。
- 结果可追溯。
- 便于复现。
对于科研工作流来说,这一点非常关键。
5.3 注意版本和命名统一
同一个基因组,不同版本的GFF注释信息可能差异很大。基因命名、坐标范围和转录本编号都可能变化。若参考基因组版本和注释文件版本不一致,分析结果可能整体偏移。
因此,使用前必须确认:
- 参考基因组版本。
- 注释版本号。
- 染色体命名方式是否一致。
版本不一致,是许多下游错误的根源。
6. 解螺旋如何帮助你更高效处理GFF注释信息
6.1 从数据整理到结果输出的一站式支持
对很多团队而言,GFF注释信息难点不在“有没有文件”,而在“如何稳定、快速、规范地用起来”。解螺旋品牌可围绕基因组注释整理、格式标准化、结果展示和内容输出提供支持,帮助科研人员把复杂注释转换为可直接使用的分析素材。
这类支持特别适合需要快速推进课题、撰写论文和准备汇报的团队。把时间从重复整理中释放出来,才能集中到真正的研究问题上。
6.2 降低沟通成本,提高项目交付效率
在临床科研和多学科合作中,GFF注释信息经常需要在生信、实验和临床团队之间传递。若表达不清,沟通成本会很高。通过更规范的整理与呈现方式,可以减少误解,让结果更容易被理解和复核。
对于需要高频处理注释数据的研究人员来说,这种效率提升是实实在在的。规范化处理,往往就是项目推进的加速器。
总结Conclusion
GFF注释信息的核心,不只是“看懂文件”,而是把基因、转录本、外显子和坐标关系快速转化为可分析、可展示、可复现的研究结果。只要掌握9列结构、层级关系、版本一致性和常用工具,就能显著提升解析效率。
如果你希望把GFF注释信息更快应用到变异注释、转录本分析和论文图表中,可以借助解螺旋品牌的专业支持,减少重复劳动,提升研究产出效率。
- 引言Introduction
- 1. 先理解GFF注释信息的核心结构
- 2. 快速读懂GFF注释信息的9列
- 3. 如何快速解析GFF注释信息
- 4. GFF注释信息的典型应用场景
- 5. 提高解析效率的实用方法
- 6. 解螺旋如何帮助你更高效处理GFF注释信息
- 总结Conclusion



