引言Introduction
DNA-seq数据量大、噪声多、文件格式复杂。很多医学生、医生和科研人员,拿到原始测序结果后,最难的不是“有没有变异”,而是“如何快速、规范地把变异挖出来 ”。DNA-seq数据的关键,在于先清洗、再注释、再筛选,减少无效计算,提高结果可解释性。
1. 先把DNA-seq数据整理到可分析状态
1.1 原始文件要先统一成标准对象
在正式挖掘变异前,第一步不是直接看结果,而是先处理输入数据。上游流程中提到,可使用 import 函数导入GTF文件,再转成数据框,提取基因或注释信息。这个思路对DNA-seq数据同样重要。先把数据整理成可重复、可调用的标准格式,后续分析才稳定。
如果数据量较大,读取过程可能较慢。这是正常现象。尤其是包含大量染色体区间、基因注释或样本信息时,建议先保存中间对象。这样下次分析时直接加载,可明显节省时间。
1.2 清洗和去重决定后续效率
DNA-seq数据的“快速”,本质上来自前处理效率。很多冗余字段、重复注释、无关染色体信息,都会增加计算负担。上游知识库中提到,在基因注释时会先过滤常染色体和XY染色体相关信息,再提取关键字段并去重,最后得到更干净的数据集。
对于变异挖掘,建议重点做三件事:
- 统一样本命名和参考版本。
- 去除重复记录和无效条目。
- 保留后续注释真正需要的字段。
数据越干净,后续变异识别越快,误差也越小。
2. 用合适的方法缩小搜索范围
2.1 变异挖掘前,先做信息压缩
DNA-seq数据的核心问题不是“数据少”,而是“信息密度高”。如果一开始就对全部位点逐一扫描,计算成本会很高。上游内容在单细胞分析中提到,会先评估基因变异性,再挑选变异程度较高的前2000个基因,以降低计算量。这个原则同样适合DNA-seq数据分析。
对于变异挖掘,思路可以类比为先聚焦高价值区域,例如:
- 重点基因区域。
- 已知热点位点。
- 临床相关候选区域。
- 质量更高、覆盖更充分的片段。
先缩小范围,再深入挖掘,是提高效率的关键。
2.2 参考版本一致,结果才可靠
DNA-seq数据分析中,参考基因组版本和注释版本必须统一。上游知识库指出,使用最新版本或旧版本注释时,匹配结果会不同,有些少见或新基因类型可能无法完全对应,但通常不影响核心分析。
这提醒我们,变异挖掘时要关注:
- 参考基因组版本是否一致。
- 注释文件版本是否匹配。
- 样本ID是否和元数据对应。
- 坐标系统是否统一。
如果版本不一致,后续筛出的变异可能看起来很多,但可信度会下降。快速不等于随意,标准化才是效率的前提。
3. 变异识别要结合统计和可视化
3.1 先看分布,再看异常
在变异挖掘中,直接报结果往往不够。更稳妥的做法,是先看总体分布,再找异常点。上游流程中提到,先观察平均表达量和变异系数,再用拟合曲线挑选高变基因。这种“先分布、后筛选”的方法,能够避免被噪声误导。
对DNA-seq数据来说,可以优先关注:
- 变异频率较高的位点。
- 质量分值较稳定的区域。
- 覆盖深度足够的区域。
- 与临床表型相关的候选变异。
统计筛选的价值,不是替代判断,而是帮助你更快锁定重点。
3.2 可视化能明显提高判断效率
上游知识库中,降维分析用到了PCA、IRLBA和t-SNE,并通过图形比较不同方法的聚类效果。虽然这是单细胞分析场景,但方法论很清晰:图比表更容易发现结构,尤其适合快速定位异常模式。
在DNA-seq数据中,可视化同样重要。常见图形包括:
- 变异位点分布图。
- 覆盖深度图。
- 火山图式筛选图。
- 样本间差异图。
- 聚类或相似性热图。
通过图形,研究者可以更快判断哪些样本需要复核,哪些变异值得优先深入。对于临床和科研场景,这一步能明显减少无效工作。
4. 用聚类和统计检验提升变异挖掘精度
4.1 聚类能帮助发现样本分层
上游知识库中,使用层次聚类和动态剪切术对细胞进行再次聚类,最终得到8类细胞,并通过颜色区分不同标签。这个逻辑在DNA-seq数据中同样适用。样本之间常常存在分层,聚类有助于把相似样本归在一起,进一步发现共性变异。
常见用途包括:
- 区分不同亚型样本。
- 识别批次效应。
- 发现异常样本。
- 提高候选变异的可解释性。
当样本先被合理分组,变异筛选的准确度通常会更高。
4.2 统计检验帮助筛掉偶然波动
上游内容提到,差异基因筛选会结合t检验、威尔逊秩和检验和二项分布,并输出P值、校正后P值、log FC等指标。DNA-seq数据挖掘变异时,也应尽量保留统计学框架,而不是只看“有没有变化”。
建议重点关注:
- 原始P值。
- 多重检验校正后的P值。
- 效应量或变异幅度。
- 样本间一致性。
只有统计显著、幅度明确、重复性较好的变异,才更值得进入后续验证。
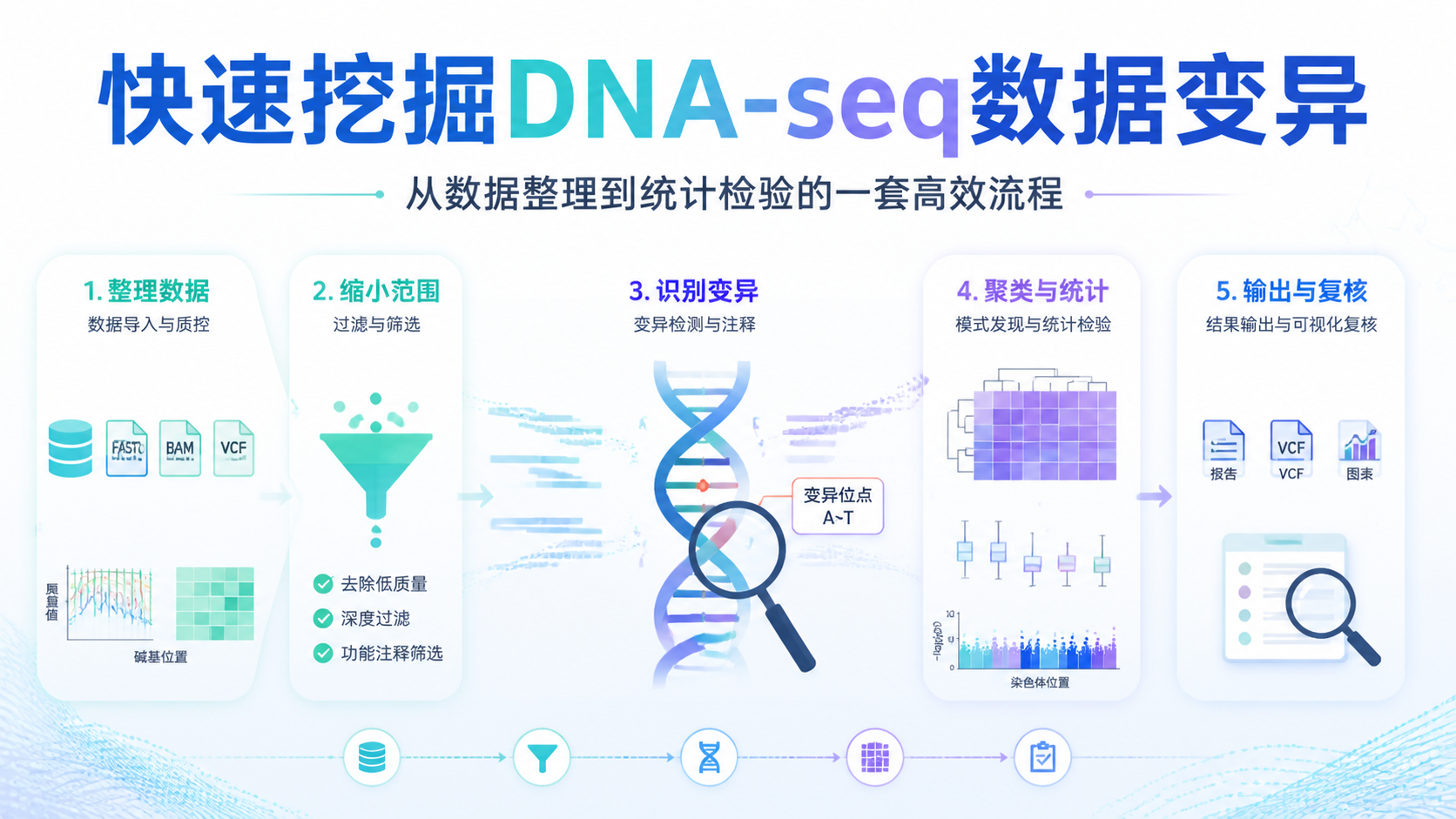
5. 快速挖掘DNA-seq数据变异的实用步骤
5.1 一套高效流程更适合科研和临床
结合上游知识库中的思路,可以把DNA-seq数据的快速变异挖掘概括为以下流程:
- 导入原始数据和注释文件。
- 统一参考版本与样本命名。
- 清洗、过滤、去重。
- 提取高价值区域或候选位点。
- 进行统计筛选和可视化。
- 输出标准化结果文件,便于复核和分享。
这套流程的优势在于清晰、可重复、易追踪。对于科研人员来说,便于复现。对于临床研究者来说,便于审阅。对于医学生来说,便于建立完整分析思维。
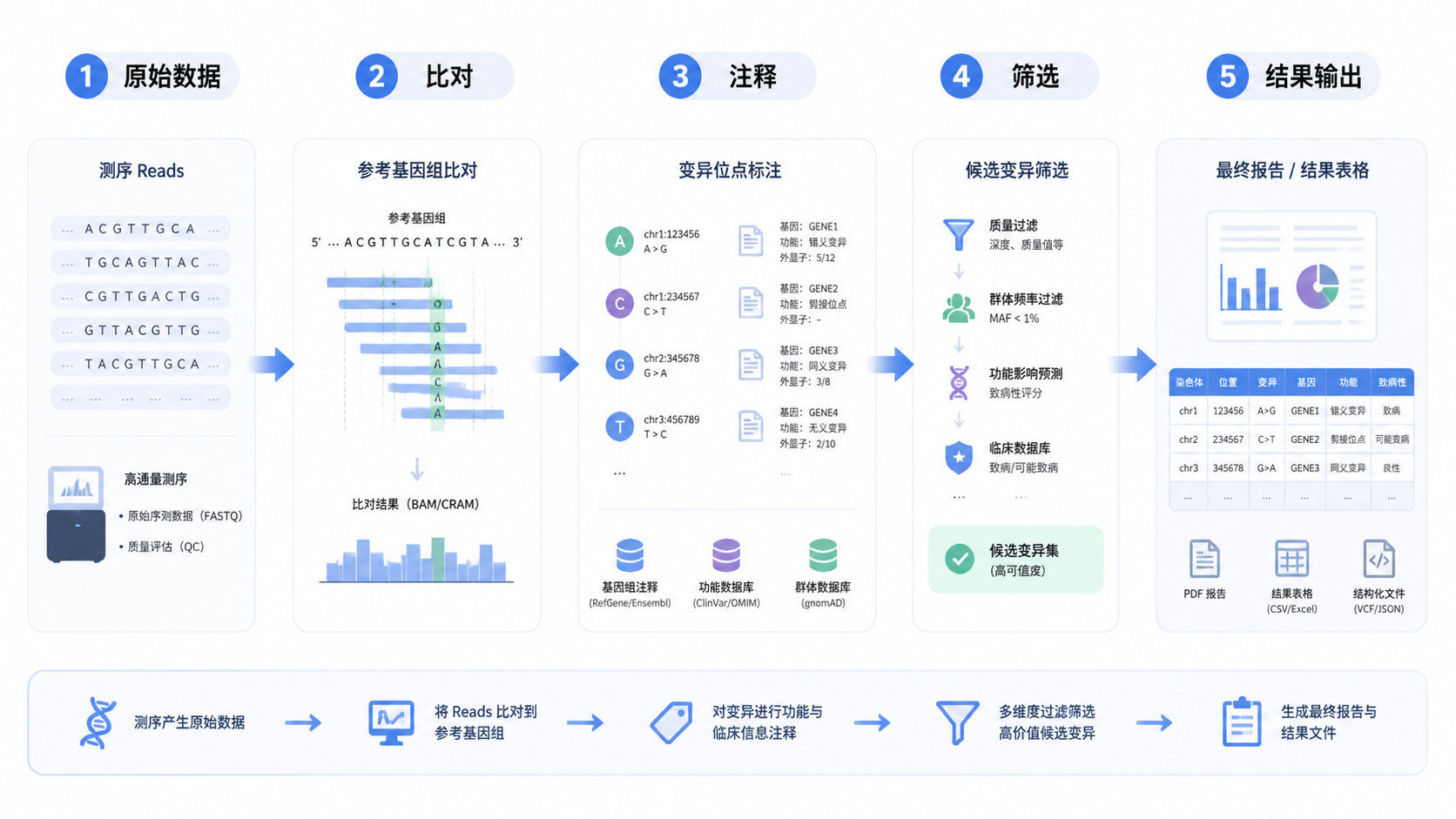
5.2 结果输出要便于复核
上游知识库强调,结果会保存到文件中,包含top排名、P值、校正后的P值、排名指标、log FC和统计量。DNA-seq数据变异分析也应如此。输出结果至少要包含:
- 变异位点。
- 基因注释信息。
- 参考与替代等位基因。
- 质量指标。
- 统计检验结果。
- 筛选阈值说明。
没有完整结果表,就很难进行后续验证和论文写作。
总结Conclusion
DNA-seq数据要想快速挖掘变异,关键不是盲目加快步骤,而是先把流程标准化。先清洗、再统一版本、再缩小范围、再做统计筛选和可视化,才能兼顾速度和可靠性。对于医学生、医生和科研人员来说,这种方法更适合实际应用,也更利于结果复现。
如果你希望更高效地完成DNA-seq数据分析,减少重复操作和格式处理时间,可以借助解螺旋 品牌提供的生信分析思路与工具支持,把复杂流程变得更清晰、更可执行。
- 引言Introduction
- 1. 先把DNA-seq数据整理到可分析状态
- 2. 用合适的方法缩小搜索范围
- 3. 变异识别要结合统计和可视化
- 4. 用聚类和统计检验提升变异挖掘精度
- 5. 快速挖掘DNA-seq数据变异的实用步骤
- 总结Conclusion