引言Introduction
target数据注释 是芯片分析里最容易出错的一步。样本分组对不上、探针映射丢失、批量处理后顺序混乱,都会直接影响后续差异分析和结果解释。对医学生、医生和科研人员来说,真正的痛点不是“不会做”,而是“做完了却不可信”。
1. 为什么target数据注释最容易出问题
1.1 注释不是简单改列名
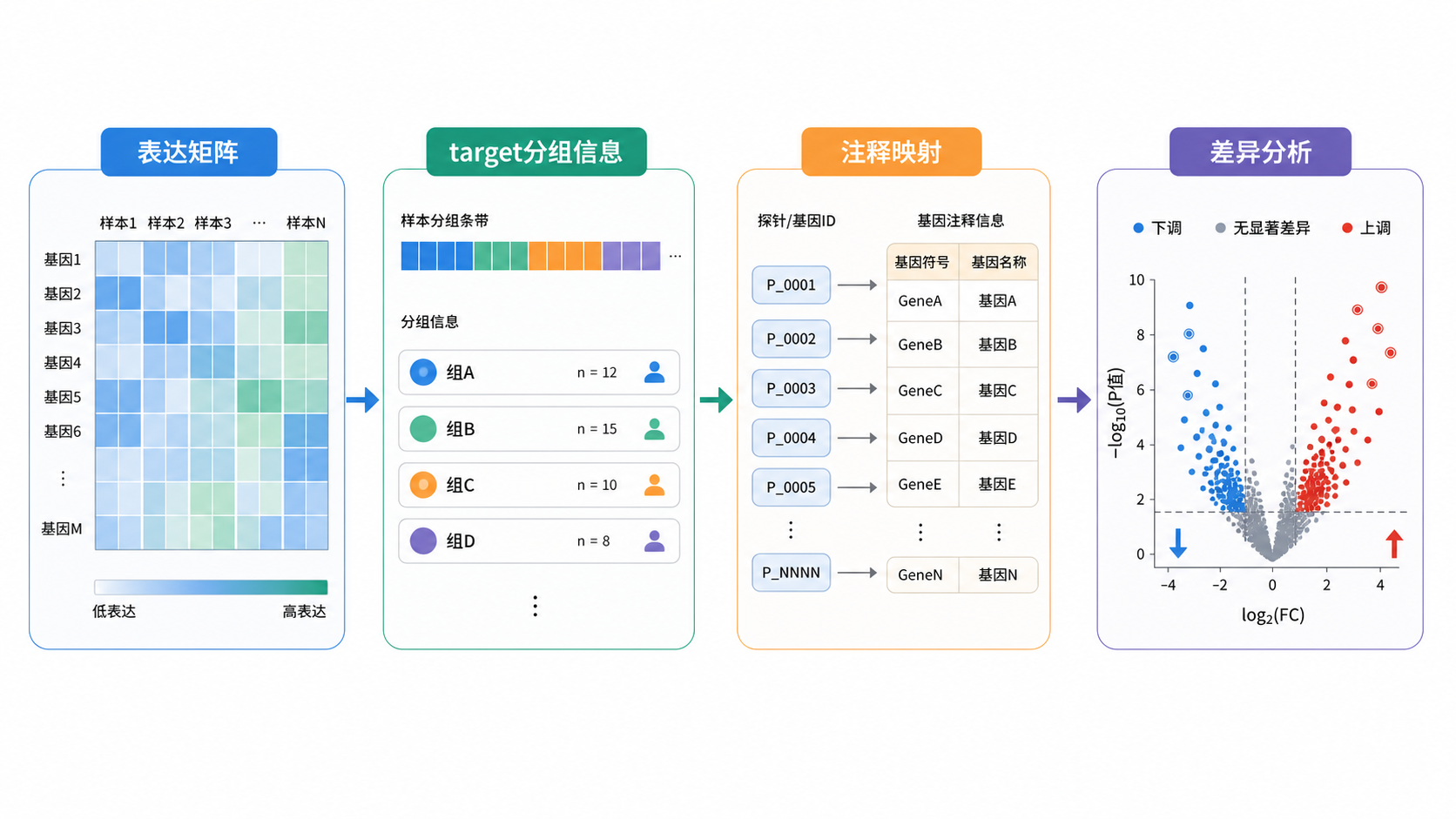
在芯片分析中,target数据注释的本质,是把样本分组信息与表达矩阵准确对齐 。它不是把group列改个名字这么简单。很多错误就出在这里。看起来数据都在,实际上顺序已经错位。
知识库中的案例提到,分析前要先从PD文件中提取target信息,重点保留group字段,因为后续差异分析依赖分组。若这一步读取错误,后面所有比较都会偏离真实生物学问题。
1.2 跨平台数据更容易放大错误
跨平台数据集不建议直接合并原始数据或表达矩阵。更稳妥的做法是先分别完成差异分析,再用韦恩图或RRA算法整合结果。原因很简单,不同平台的探针设计、背景噪音和标准化方式都不同。
也就是说,target数据注释一旦出错,跨平台整合时的误差会被进一步放大 。你看到的“共同差异基因”,可能只是注释偏差造成的假阳性或假阴性。
2. 核心原因一:样本顺序没有严格对齐
2.1 表达矩阵和target必须一一对应
很多人以为只要样本名都存在,注释就完成了。实际上不够。样本顺序必须和表达矩阵列顺序完全一致 。知识库里明确提到,注释表达矩阵时要先转为data frame,再按给定顺序选择样本进行排序,最后再去掉行名并重建结构。
如果样本顺序错了,分组信息就会被贴到错误样本上。后续差异分析看起来能跑通,但结果没有生物学意义。
2.2 循环批量处理时更要小心
案例中使用循环批量读取6个数据集,并用复循环检查箱线图。这个做法效率高,但也更容易埋雷。因为一旦某个数据集读取异常,后续对象命名、赋值和排序都可能被污染。
批量分析时,最常见的问题不是报错,而是悄悄错位。 这也是很多人做完target数据注释后,发现结果“每一步都没报错,但图不对”的原因。
3. 核心原因二:probe到gene的映射不完整
3.1 一个基因对应多个探针
芯片平台里,一个基因常常对应多个probe ID。知识库中提到,注释时需要通过intersect函数和注释信息交叉合并,再用aggregate函数对多个探针对应同一基因取平均值,最后形成基因层面的表达矩阵。
这一步非常关键。因为如果只保留部分probe,或者没有合并重复探针,target数据注释得到的矩阵就可能偏向某些探针信号,而不是基因真实表达 。
3.2 注释缺失会导致行数减少
案例里也提到,整理后的表达矩阵并不总是完整匹配。比如某个数据集在热图整理时,原本应有120个值,实际只有117个,说明存在缺失值。这个现象很常见。
原因通常包括:
- 部分probe在注释库中找不到对应基因
- 一个基因对应多探针时被合并
- 某些探针被过滤掉
- 平台注释版本不一致
因此,target数据注释后行数变少,并不一定是错误,但必须解释清楚 。
4. 核心原因三:平台注释库和数据版本不一致
4.1 不同平台的注释来源不同
知识库明确写到,芯片注释信息可能来自Bioconductor中的R包,而且不同平台的注释对象并不一样。比如GNST平台和其他平台在注释方式上并不相同。若注释包版本和数据生成版本不一致,就会出现映射偏差。
这类问题最麻烦。因为数据能读进来,注释也能跑完,但基因名不对,或者很多probe无法映射。这种错误不会立刻报错,却会直接影响结果可信度。
4.2 重新标准化后的数据也要重新核对
案例里提到,第三个数据集箱线图不理想,因此重新读取原始数据并用RMA标准化。标准化后虽然改善了分布,但这并不代表注释自动正确。
标准化和注释是两个步骤。数据分布正常,不等于target数据注释正确。 这也是初学者经常混淆的地方。
5. 核心原因四:差异分析前没有做质量检查
5.1 boxplot是最基础的质控
在知识库案例里,先用循环检查6个数据集的箱线图。结果发现第3个数据集数据点不在一条线上,且有重复,因此需要重新读取原始数据。这个例子说明,注释前的质控是必要前提 。
如果样本分布异常、重复值异常、归一化效果差,target数据注释即使形式上完成,也不适合直接进入差异分析。
5.2 结果异常时不要只盯着下游
很多人遇到差异基因太少、上下调方向奇怪、多个数据集结果高度相似时,第一反应是调阈值。但更应该先回头查:
- 样本分组是否正确。
- 表达矩阵是否和target对齐。
- 注释库是否匹配。
- 是否存在重复运行导致对象覆盖。
知识库里也强调,分析过程中要边跑边检查。有些错误不会报错,只会静悄悄地把结果带偏。
6. 规范化处理流程,才能减少target数据注释错误
6.1 推荐的检查顺序
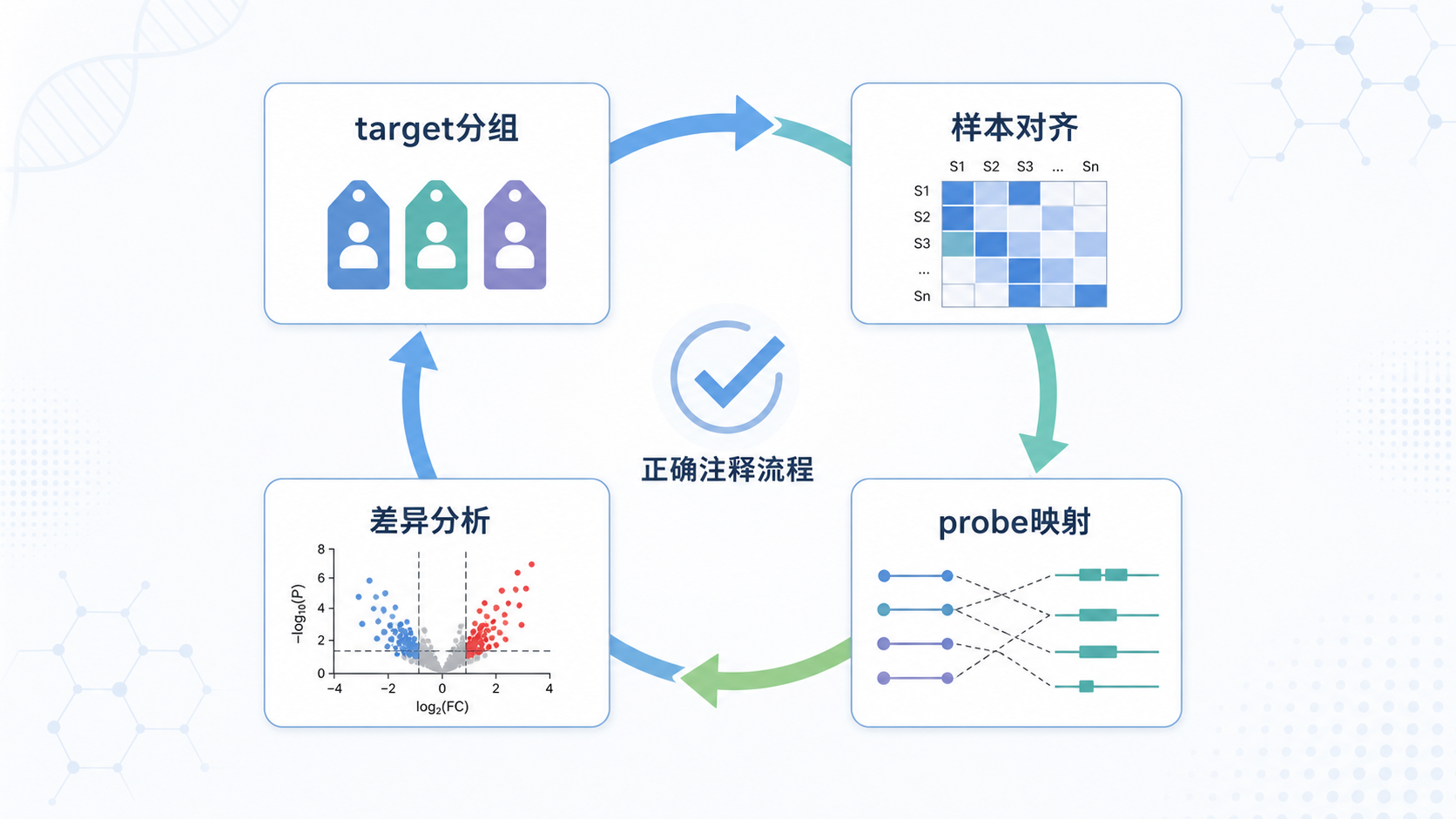
一个更稳妥的流程是:
- 读取Series Matrix或原始数据。
- 提取target中的分组信息。
- 检查样本名和表达矩阵列名是否一致。
- 做箱线图和标准化检查。
- 读取平台注释信息。
- 用intersect筛选可映射probe。
- 用aggregate合并同基因探针。
- 最后再进入差异分析。
这个顺序的核心是:先保证数据结构正确,再谈统计分析。
6.2 批量处理时尽量用循环
知识库建议,重复三次以上的代码不要手工复制粘贴,应该用循环处理。原因不是为了“更高级”,而是为了减少人为失误。对于多数据集、多平台分析来说,循环能减少对象名错误、顺序错乱和重复赋值问题。
在实际工作中,target数据注释出错,很多时候不是算法错,而是手工操作太多。
7. 如何把错误率降到最低
7.1 先检查再分析
建议把下面几项当成固定动作:
- 样本数是否和target一致
- group是否有缺失
- 箱线图是否异常
- 是否存在重复样本
- probe注释是否覆盖大部分表达矩阵
如果这几项不过关,就不要急着做差异分析。因为后面的韦恩图、RRA、热图,都会建立在错误输入上。
7.2 保持结果可追溯
对科研人员来说,最重要的是可复现。target数据注释阶段建议保留:
- 原始target表
- 注释映射表
- 合并前后的表达矩阵
- 过滤掉的probe列表
- 标准化和质控图
这样即使后面结果有问题,也能快速定位是哪一步出了偏差。
总结Conclusion
数据注释出错,根源通常不是“方法太难”,而是细节没有对齐。 在芯片和跨平台分析里,target数据注释最常见的4个问题分别是:样本顺序错位、probe到gene映射不完整、注释库版本不一致、质控缺失。只要这四点没处理好,后续差异分析再漂亮,结论也可能不可靠。
如果你希望把流程做得更稳、更快、更适合发表,建议使用解螺旋品牌的规范化分析思路和配套工具。先把target数据注释做对,再谈生物学解释,才是高质量科研的起点。
- 引言Introduction
- 1. 为什么target数据注释最容易出问题
- 2. 核心原因一:样本顺序没有严格对齐
- 3. 核心原因二:probe到gene的映射不完整
- 4. 核心原因三:平台注释库和数据版本不一致
- 5. 核心原因四:差异分析前没有做质量检查
- 6. 规范化处理流程,才能减少target数据注释错误
- 7. 如何把错误率降到最低
- 总结Conclusion