引言Introduction
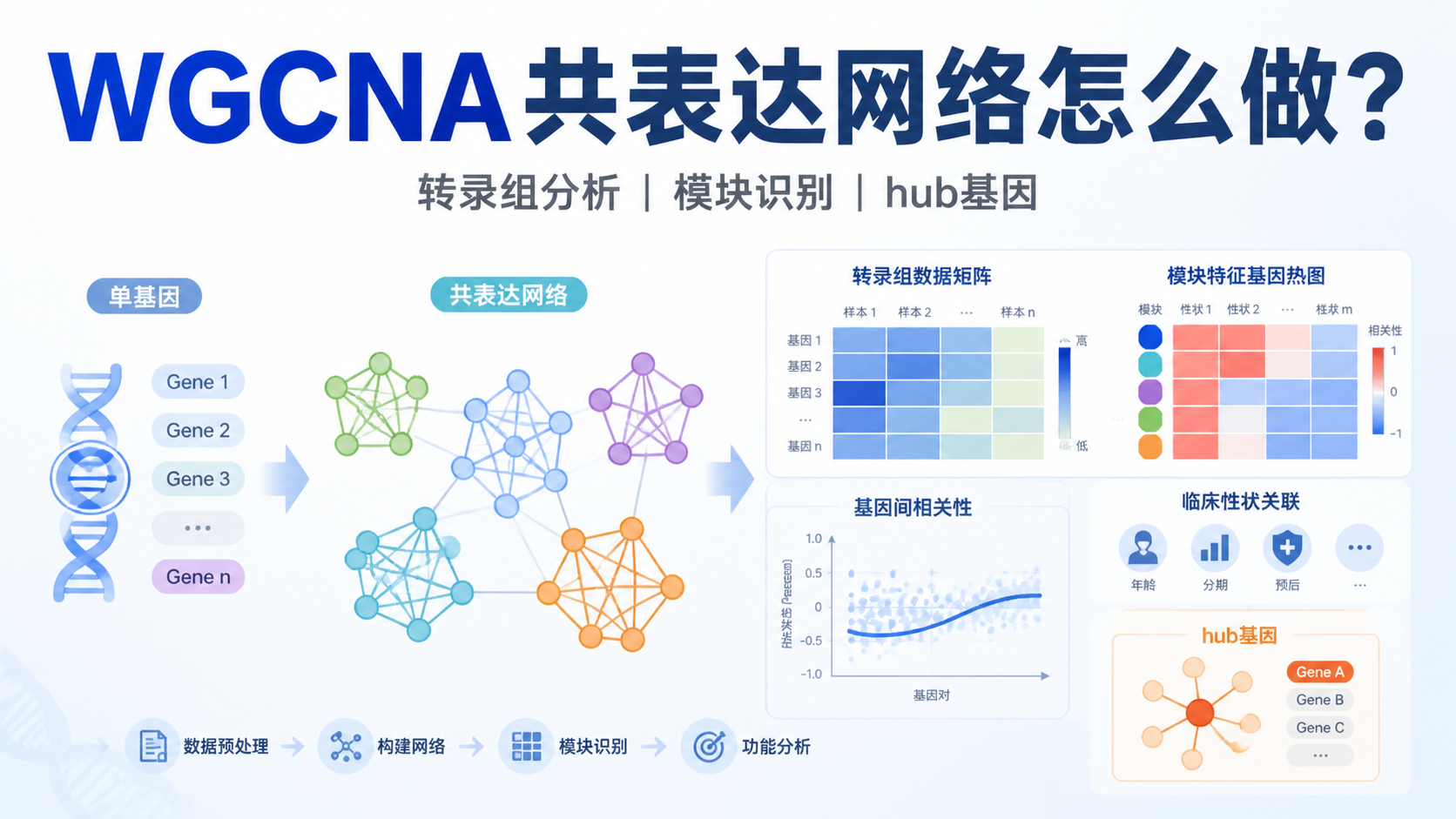
加权基因共表达网络 常用于转录组数据中,帮助研究者从海量基因里找出协同变化的模块,再进一步关联表型、筛选核心基因。对医学生、医生和科研人员来说,真正的痛点不是“有没有数据”,而是“怎么把表达矩阵变成可解释的生物学结论”。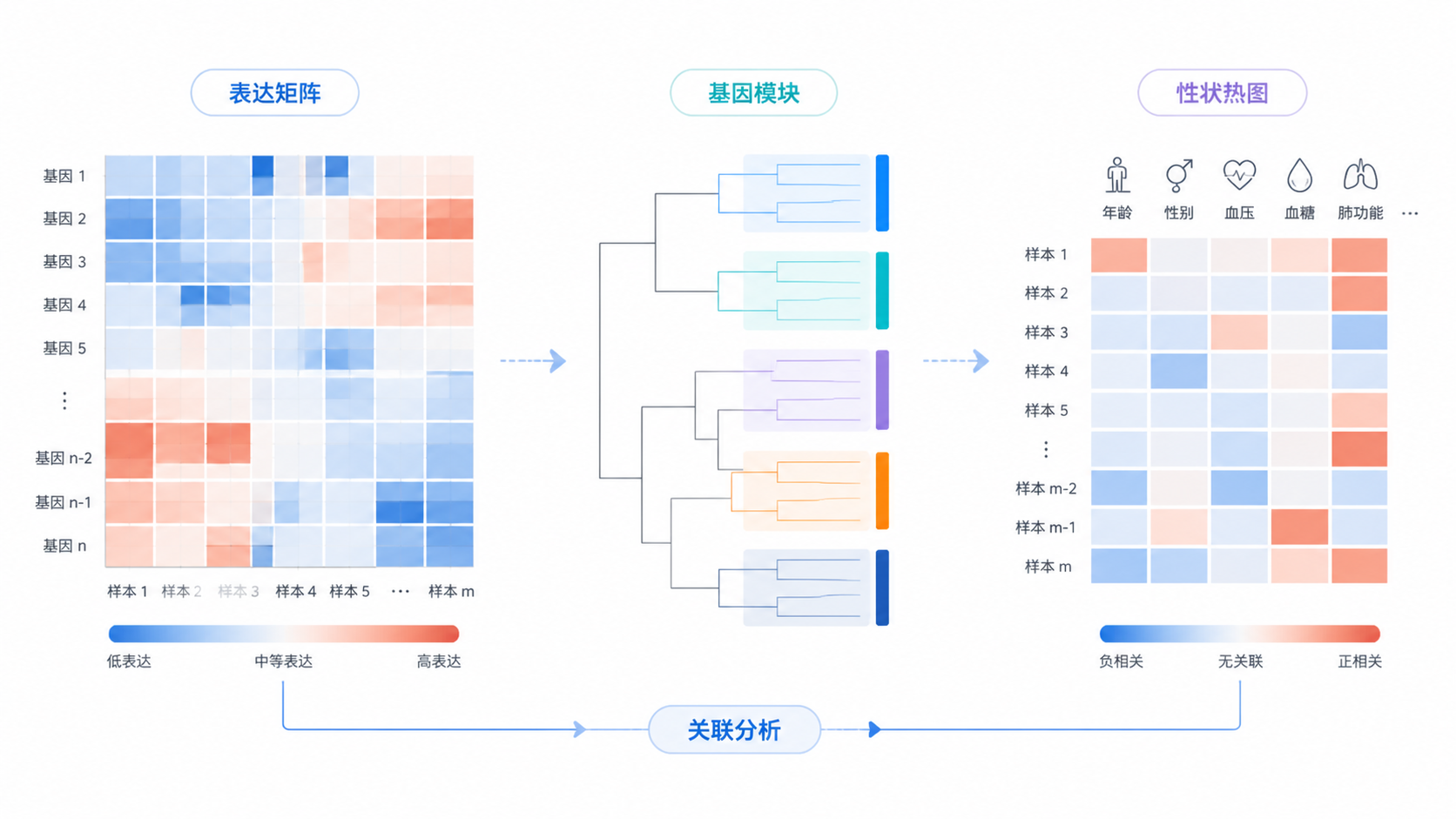
1. 加权基因共表达网络的核心用途
1.1 从单基因思维转向模块思维
加权基因共表达网络的目标,不是盯住单个差异基因,而是识别表达模式相近的基因模块 。这些模块往往反映同一生物过程或同一调控轴。相比只看DEG列表,这种方式更适合复杂疾病、发育过程和感染应答。
在课程知识库中,WGCNA的核心目的包括三点。
- 寻找协同表达的基因模块。
- 探索基因网络与表型的关联。
- 进一步锁定核心基因。
1.2 适合什么样的转录组场景
加权基因共表达网络 更适合样本量相对充足、表型信息明确的数据。知识库明确提到,通常建议有5组或15个样品以上 ,样本越多,模块稳定性通常越好。
常见应用场景包括。
- 不同器官或组织的发育调控。
- 同一组织不同发育阶段比较。
- 病原菌侵染后的时间序列表达响应。
- 与临床分组相关的分子模块筛选。
1.3 为什么它适合转录组
转录组数据本质上是高维数据。单纯做差异分析,容易得到大量基因,但难以直接解释通路层面的协同变化。加权基因共表达网络 通过基因间相关性构网,把“离散基因”组织成“功能模块”,更利于后续做机制阐释和标志物筛选。
2. 转录组里怎么搭建加权基因共表达网络
2.1 输入数据要先准备好
加权基因共表达网络 既适用于测序数据,也适用于芯片数据。
转录组分析时,建议使用RPKM、TPM或其他归一化后的表达量 。如果是芯片数据,则使用常规归一化矩阵。
知识库中的示例数据包含407例样本、8840个基因 。这说明WGCNA不是针对少量样本的简单聚类,而是面向较大规模表达矩阵的网络分析。
2.2 先做数据检查,再做网络构建
第一步是数据转置和质量检查。课程中使用goodSamplesGenes函数检查缺失条目。
如果返回TRUE,说明没有低质量样品或基因。这个步骤很关键,因为异常值会直接影响后续模块识别。
随后要做样品聚类。
- 使用
dist函数计算样品间欧式距离。 - 通过层次聚类识别异常样品。
- 再根据剪切高度去除离群点。
知识库示例中,剪切高度设为155 ,最终保留406例样品 。这类处理能明显提高网络稳定性。
2.3 临床信息要和表达矩阵对齐
如果要做模块与表型关联,就必须准备临床信息表。
常用做法是先整理样品分组,再用intersect筛出表达矩阵和临床表中的共同样品名,保证两者一一对应。
知识库中还提到,可使用number2colors把临床信息转成颜色,再用plotDendroAndColors绘制样品聚类树和性状图。这样能直观看到样本分组是否与表达模式一致。
3. 网络构建的关键参数怎么选
3.1 软阈值决定网络是否接近无尺度特征
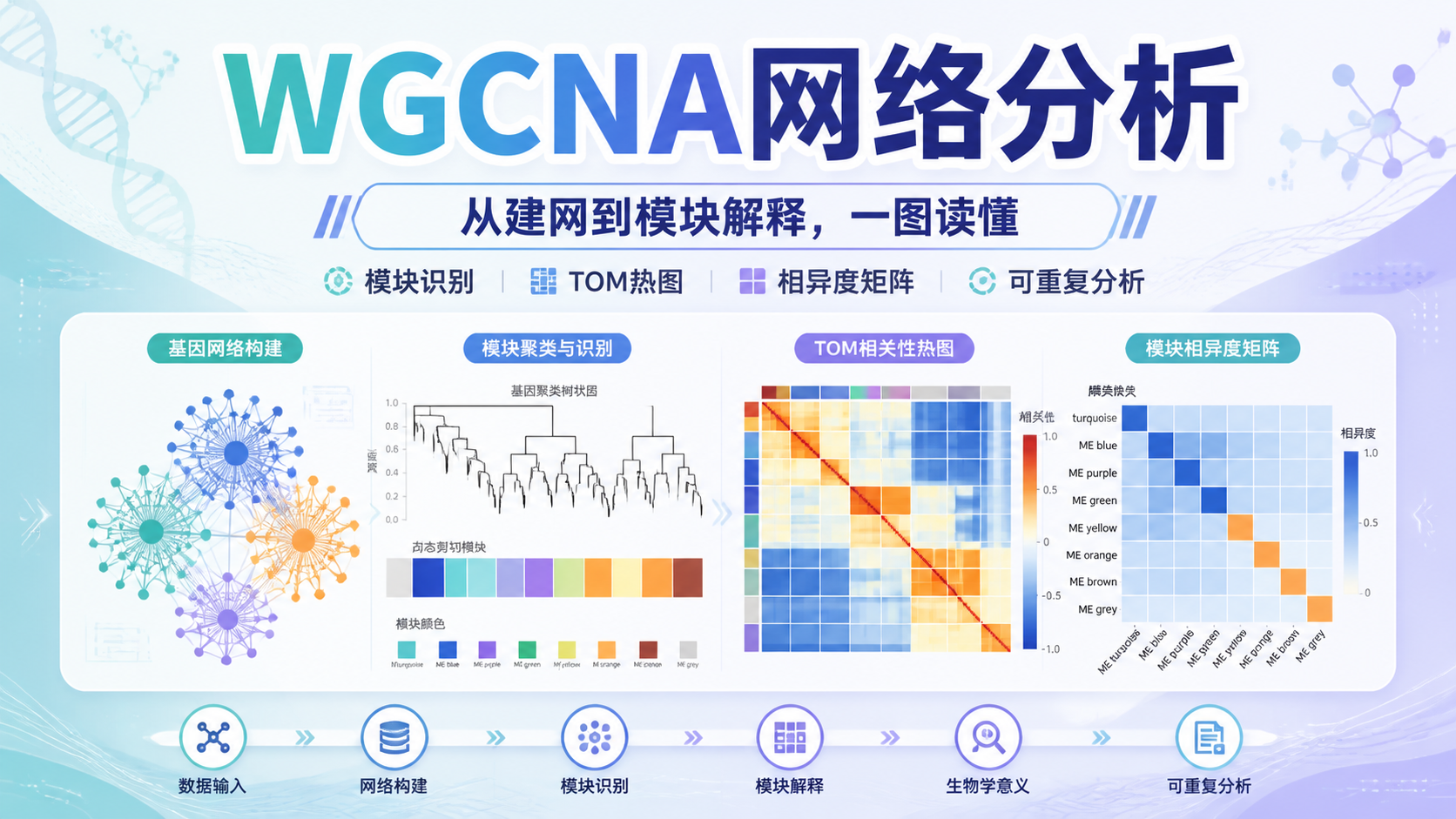
在加权基因共表达网络 里,最关键的参数之一是软阈值,也叫power。课程给出的候选范围是1到20 ,并使用pickSoftThreshold评估不同power值。
选择原则不是盲目追求最大值,而是让网络尽量符合无尺度网络 特征。知识库示例中,通常会选择3或4 作为较合适的结果。这个步骤决定后续邻接矩阵的构建质量。
3.2 从相关性到拓扑矩阵
WGCNA的基本逻辑是。
- 先计算基因表达相似性。
- 再通过软阈值加权,得到邻接矩阵。
- 进一步计算拓扑重叠矩阵,TOM。
- 最后用TOM进行基因聚类。
TOM的意义在于,它不仅考虑两个基因是否相关,还考虑它们与其他基因的共同连接情况。因此,加权基因共表达网络 比单纯相关系数网络更稳健。
3.3 模块识别靠动态剪切
在得到基因聚类树后,使用cutreeDynamic进行动态剪切,识别不同模块。知识库示例中,最终得到13个不同的基因模块 ,再用labels2colors为模块赋色。
这一步的输出,通常是后续分析的核心图之一。它告诉你,哪些基因属于同一共表达模块。
4. 如何把模块和临床性状联系起来
4.1 模块不是终点,关联表型才是重点
加权基因共表达网络 真正有价值的地方,在于模块与临床性状的相关性分析。
做法通常是计算每个模块的特征基因与表型之间的相关系数和P值,再用热图展示结果。
课程知识库明确指出,模块与临床性状相关性分析要同时看相关系数和P值 。
这能帮助你判断哪些模块更值得进入后续机制研究。
4.2 先筛模块,再筛核心基因
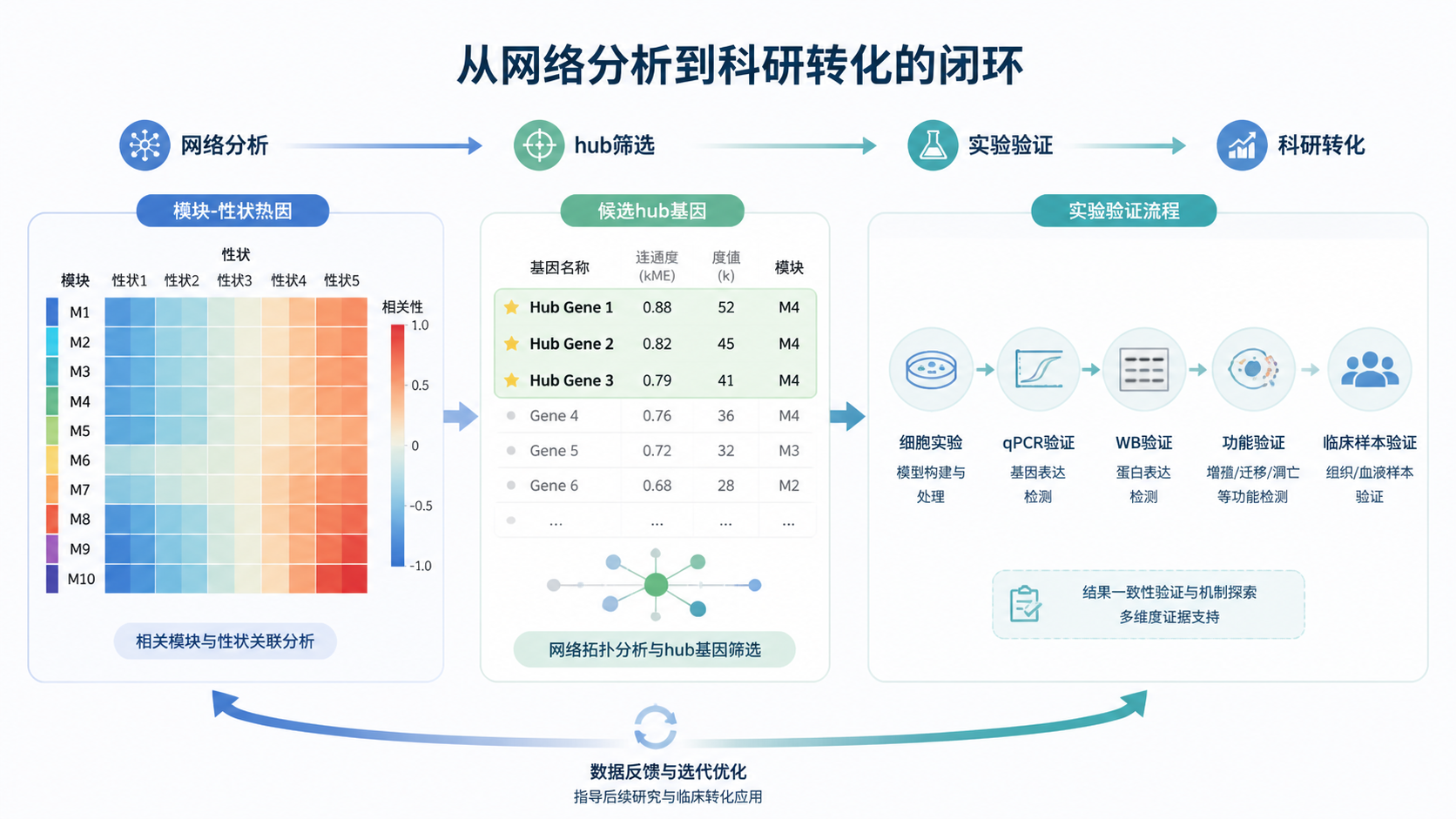
当热图显示某个模块与目标表型高度相关时,就可以进一步分析模块内基因与性状的关系。知识库中提到,还可以计算模块与基因之间的相关性矩阵,并批量输出感兴趣模块的散点图。
这一步的逻辑是。
- 先找与表型相关的模块。
- 再找模块内与表型最相关的基因。
- 最后聚焦可能的hub基因。
4.3 输出模块基因便于后续验证
课程最后一步是输出每个颜色模块的基因,选择最相关模块开展后续分析。
实际研究中,这些基因常被进一步用于GO、KEGG富集分析,或结合qPCR、蛋白实验、数据库验证进行功能确认。
对于科研人员来说,这一阶段的目标不是“画完图”,而是形成可验证的候选基因列表 。
5. 转录组中使用加权基因共表达网络的实操建议
5.1 不要直接把所有基因都扔进去
虽然官网推荐可用全部表达基因,但知识库也提醒,实际操作中通常更推荐选取变异程度较大的基因 。例如可基于方差、中位数绝对偏差等指标排序,取前5000个基因 左右。
原因很直接。
- 基因太少,网络不稳定。
- 基因太多,计算压力大。
- 低变异基因通常对模块贡献有限。
5.2 先保证样本质量,再谈生物学解释
加权基因共表达网络 对离群样本很敏感。
如果样本聚类图里存在明显异常点,应该优先处理,而不是直接进入模块分析。否则,模块和性状的相关性可能会被噪音拉低。
5.3 结果解读要避免过度推断
WGCNA给出的只是共表达关系 ,不是因果关系。
模块相关,不代表直接调控。
hub基因重要,也不等于就是致病基因。
因此,最好结合差异表达、富集分析、公开数据库和实验验证一起判断。
6. 研究者最关心的结果应该怎么看
6.1 先看模块颜色,再看性状热图
在标准流程中,研究者一般先看模块树状图,再看模块-性状热图。
如果某个模块与疾病分组、分期、时间点或组织类型显著相关,这个模块就是优先研究对象。
6.2 再看模块内基因的生物学一致性
高质量的加权基因共表达网络 模块,通常会呈现相对一致的生物学功能。
比如免疫相关模块、代谢相关模块、细胞周期相关模块,内部基因往往具有相近的功能注释。
6.3 最后锁定候选hub基因
从相关模块中筛出hub基因后,常见后续路径包括。
- 与差异基因取交集。
- 做功能富集。
- 结合临床预后分析。
- 设计实验验证表达和功能。
总结Conclusion
加权基因共表达网络 是转录组分析中非常实用的模块化方法。它能把高维表达数据转成可解释的基因模块,再进一步连接临床性状和核心基因。对医学生和科研人员来说,关键不是只会跑流程,而是理解数据清理、软阈值选择、模块识别和性状关联这四个核心环节。
如果你希望少走弯路,可以直接参考解螺旋 的标准化分析思路和课程资源,把WGCNA从“会做”提升到“做对”。加权基因共表达网络 分析的价值,最终体现在可重复、可解释、可验证。
- 引言Introduction
- 1. 加权基因共表达网络的核心用途
- 2. 转录组里怎么搭建加权基因共表达网络
- 3. 网络构建的关键参数怎么选
- 4. 如何把模块和临床性状联系起来
- 5. 转录组中使用加权基因共表达网络的实操建议
- 6. 研究者最关心的结果应该怎么看
- 总结Conclusion