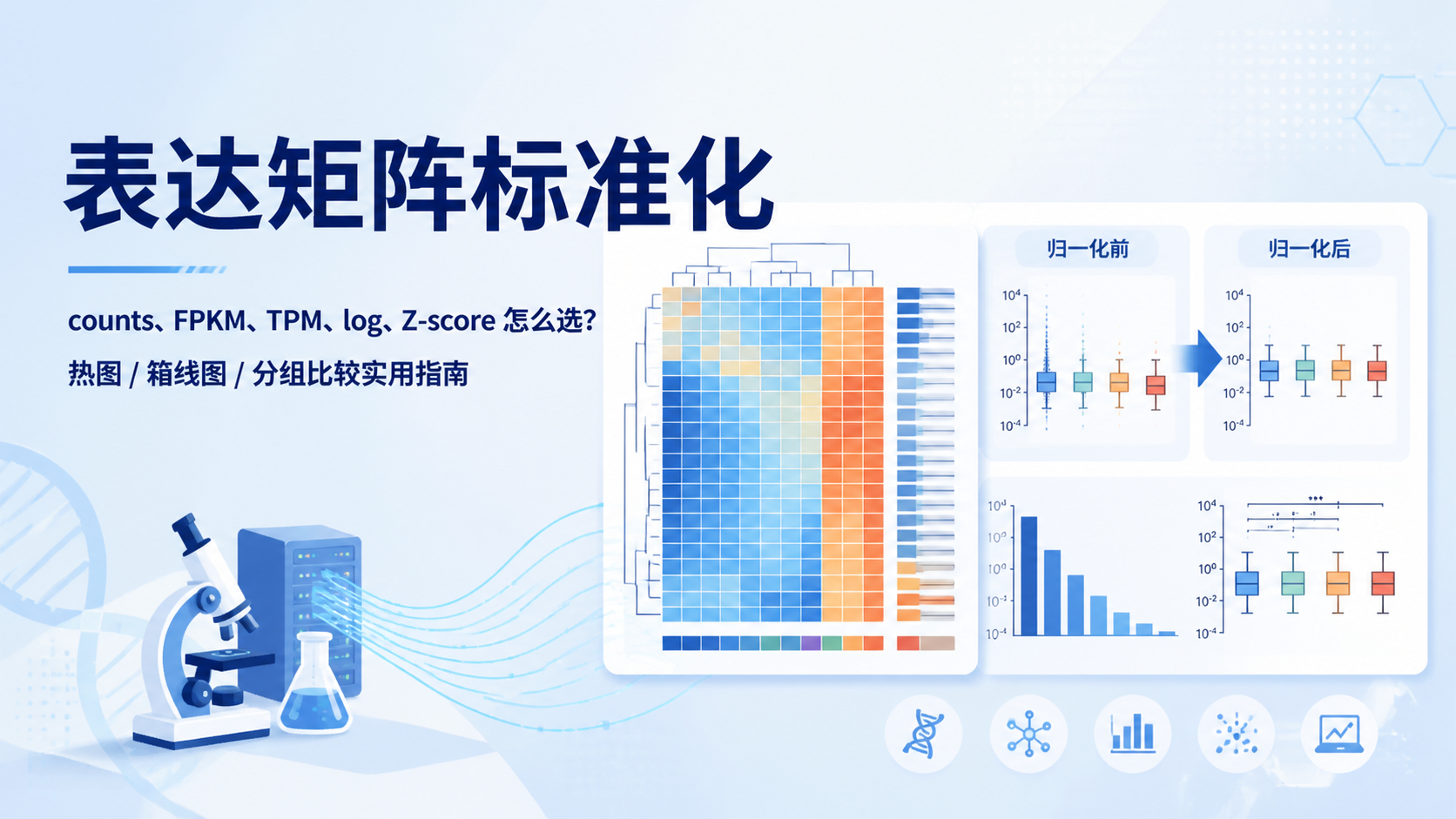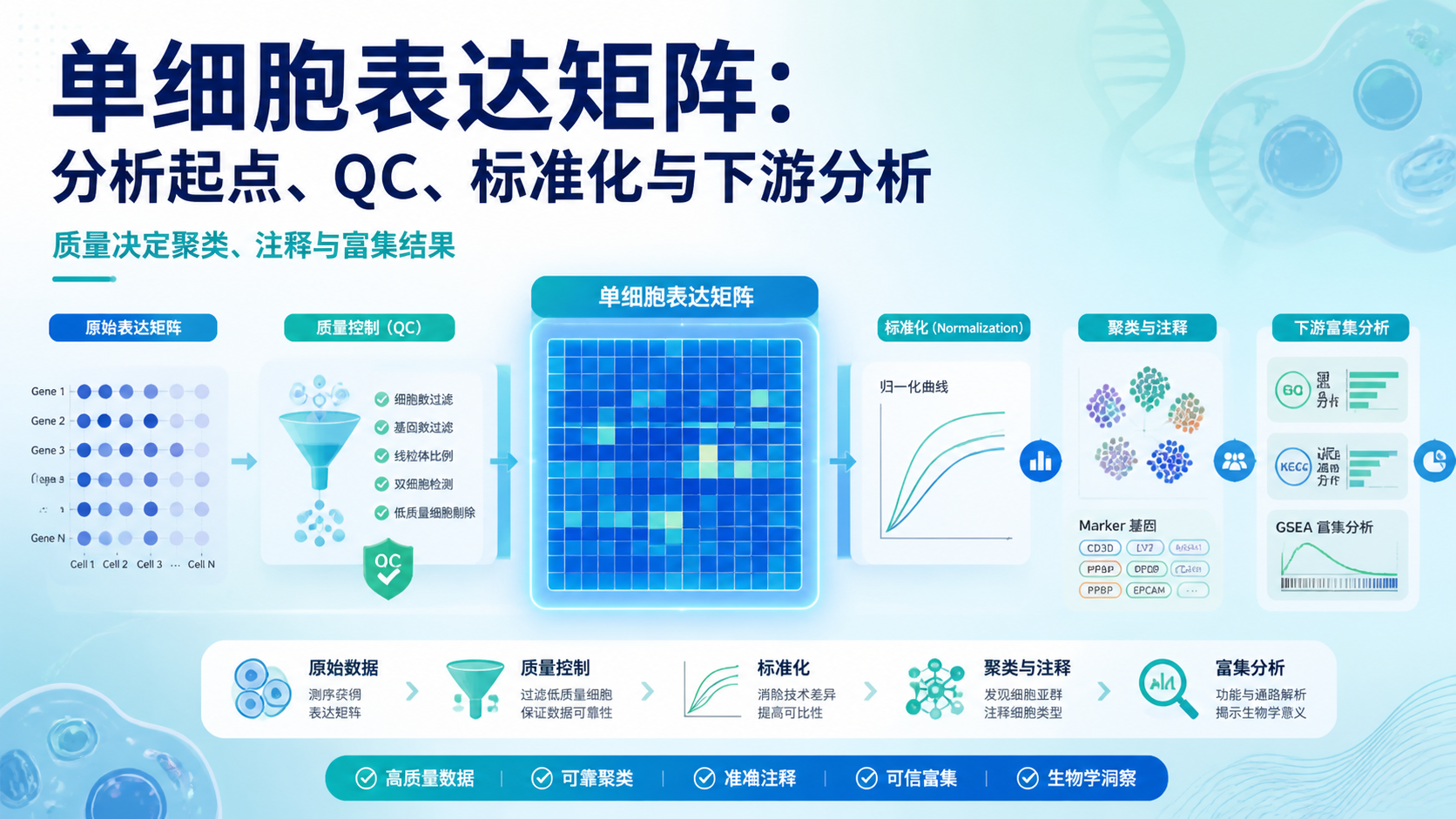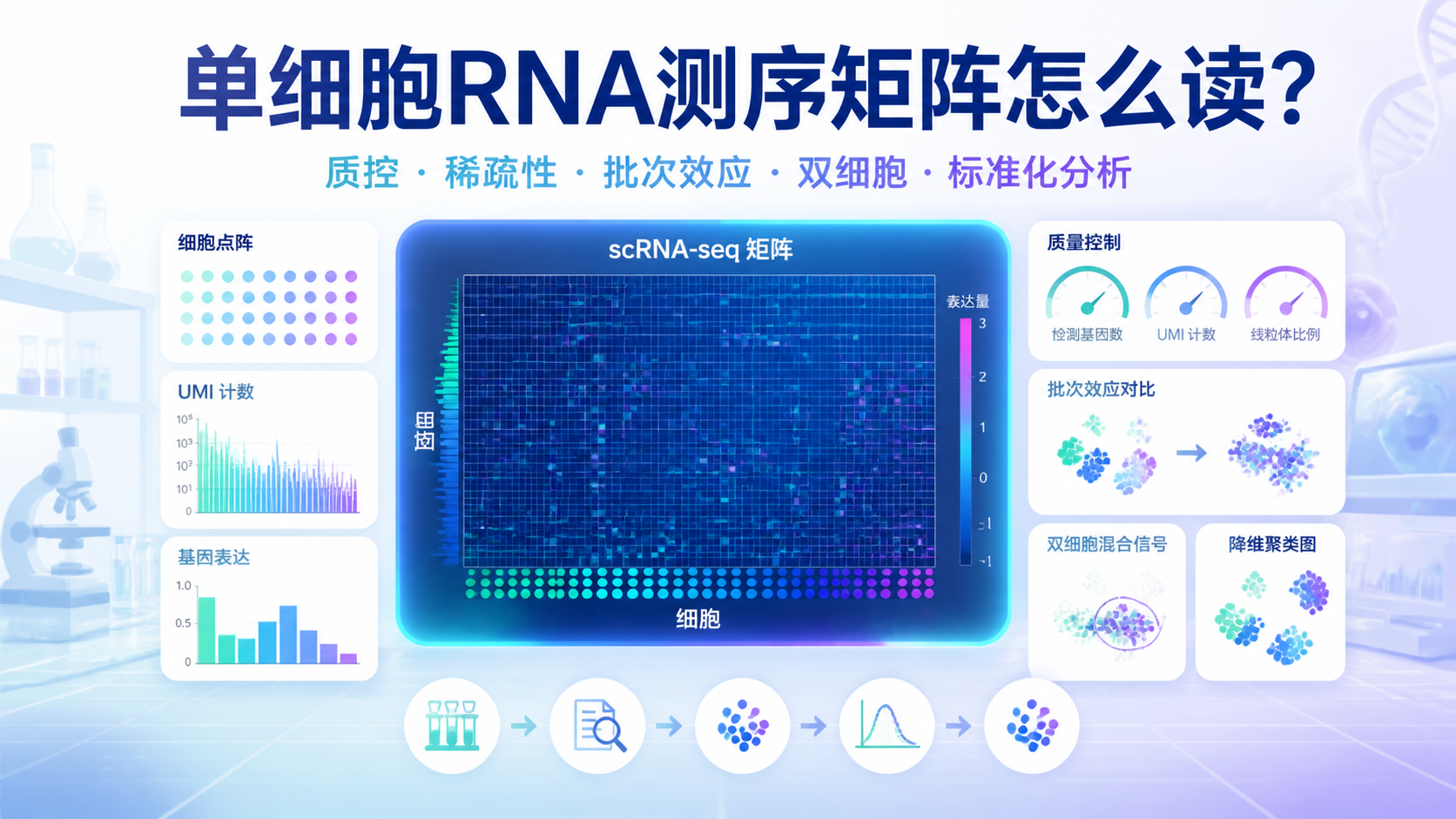
引言Introduction
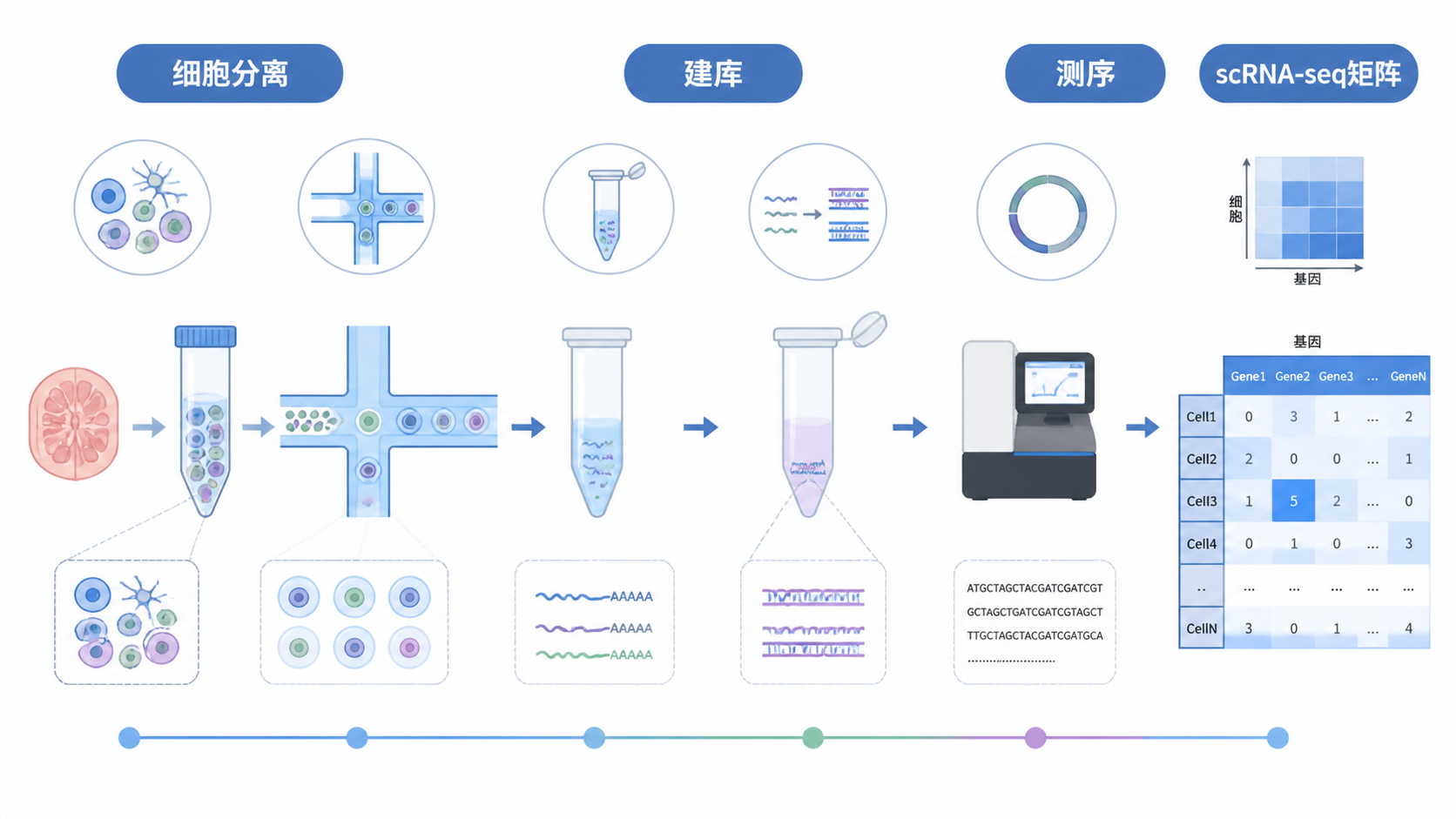
scRNA-seq矩阵是单细胞转录组分析的起点,也是最容易出错的环节之一。很多研究者拿到原始结果后,常被维度、稀疏性、批次效应和注释规则困住。如果矩阵前处理不规范,后续聚类、差异分析和细胞注释都会偏离真实生物学结论。 本文围绕 scRNA-seq矩阵,拆解5个关键要点,帮助医学生、医生和科研人员更高效地读懂数据。
1. scRNA-seq矩阵到底是什么
1.1 行、列和数值分别代表什么
scRNA-seq矩阵通常是一个基因表达矩阵。行代表基因,列代表单个细胞,数值代表该基因在该细胞中的表达量。 常见数据形式包括原始 UMI 计数、标准化表达值,或对数转换后的矩阵。
在 10x Genomics 等平台中,最常见的是稀疏矩阵格式。因为大多数基因在单个细胞里没有检测到,矩阵中会出现大量 0。这不是异常,而是单细胞数据的典型特征。
1.2 为什么它比 bulk RNA-seq 更复杂
bulk RNA-seq 反映的是群体平均表达,而 scRNA-seq矩阵记录的是单细胞层面的异质性。这意味着它既能揭示罕见细胞群,也更容易受到掉落事件、低捕获效率和环境 RNA 污染的影响。
因此,面对 scRNA-seq矩阵,不能只看表达高低,还要看数据质量、细胞数量、基因覆盖和噪音水平。只有先理解矩阵本身,后面的分析才有意义。
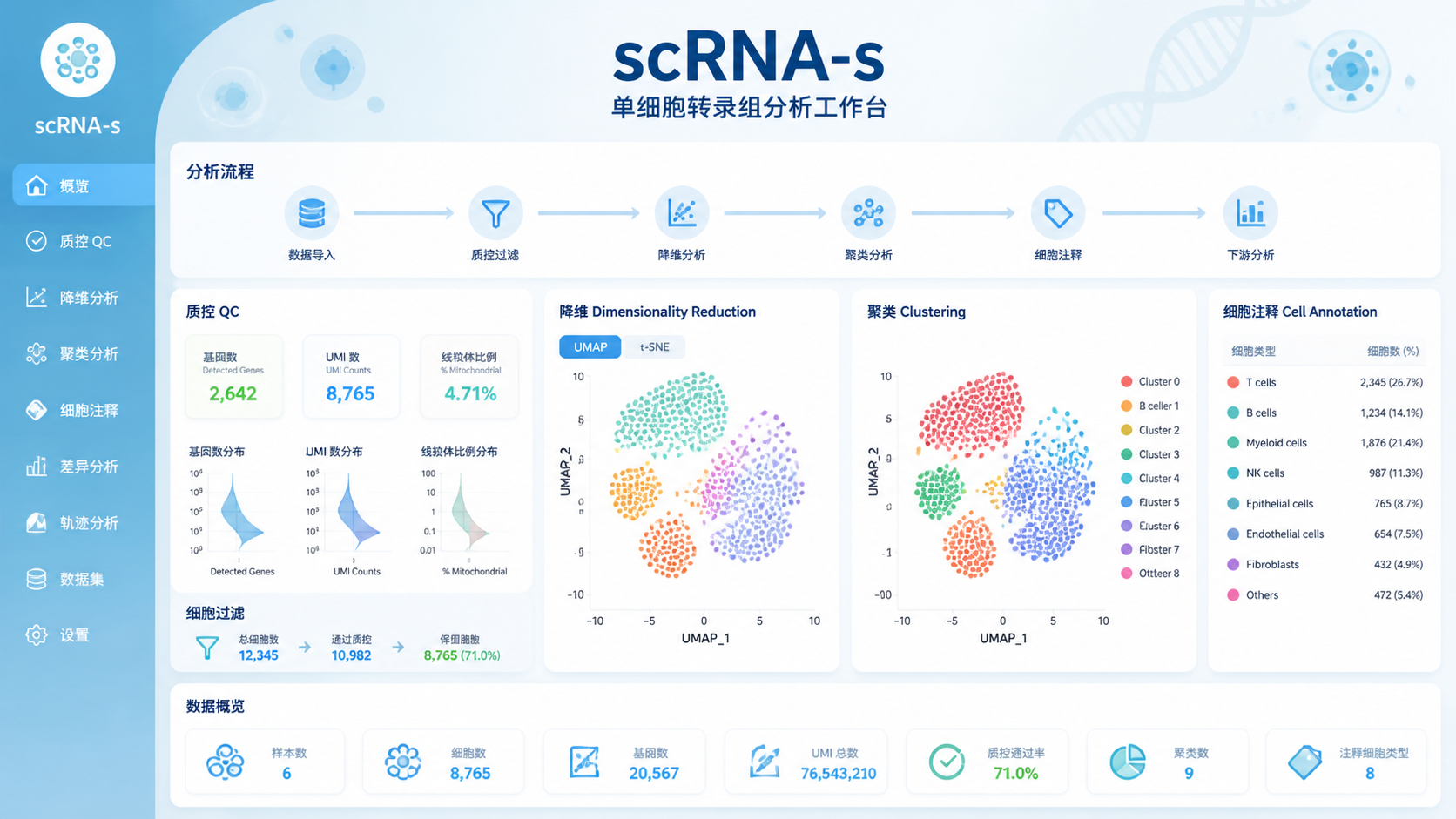
2. 解读 scRNA-seq矩阵前,先看3个核心质量指标
2.1 每个细胞检测到的基因数
每个细胞的基因检测数,常用于判断细胞质量。过低通常提示低质量细胞、空滴或破裂细胞。过高则可能是双细胞或多细胞混合。
实际分析中,研究者常结合分布图设定过滤阈值。这个阈值没有统一标准,应根据样本类型、平台和组织来源具体调整。 例如,某些组织本身转录活性高,基因数自然更多,不能机械套用别人的阈值。
2.2 每个细胞的 UMI 总数
UMI 总数反映捕获到的转录本数量。UMI 过低说明测序深度不足或细胞状态差。 UMI 过高则要警惕双细胞或异常高表达细胞。
在实际研究中,UMI 分布常与基因数联合判断。两者一起看,比单独看一个指标更稳妥。这样能减少因极端值造成的误筛。
2.3 线粒体基因比例
线粒体基因比例是单细胞质控的重要指标。通常,线粒体比例升高提示细胞应激、凋亡或破裂。高线粒体比例细胞往往不适合进入后续聚类和差异分析。
但阈值同样需要分场景处理。不同组织、疾病状态和样本处理流程,线粒体比例分布差异很大。科研人员应先看整体分布,再结合实验背景决定筛选范围。
3. scRNA-seq矩阵最容易忽视的3类问题
3.1 稀疏性不是错误,但会影响统计分析
scRNA-seq矩阵天然稀疏。大量 0 值会影响距离计算、降维和聚类结果。这也是为什么单细胞分析常需要专门的归一化、特征选择和降维方法。
如果把稀疏矩阵直接当作普通表达矩阵处理,很容易得到不稳定的聚类结构。尤其在低深度测序数据中,稀疏性会进一步放大噪音。
3.2 批次效应会扭曲真实细胞差异
不同批次、不同操作者、不同上机时间,都可能引入批次效应。它会让同一类细胞在降维图上分开,看起来像不同群体。这类假象在 scRNA-seq矩阵分析中非常常见。
因此,整合多个样本时,必须在生物学信号和技术偏差之间做平衡。过度校正会抹掉真实差异,校正不足又会保留批次噪音。关键在于根据研究目的选择合适策略。
3.3 双细胞会制造“混合表达”假象
双细胞是单细胞实验中的经典问题。两个细胞同时被捕获到同一个液滴里,形成一个“复合信号”。在 scRNA-seq矩阵中,这类细胞往往同时表达两类本不应共存的标记。
如果不做识别,双细胞会干扰细胞聚类、亚群注释和差异基因分析。对于样本量大、上样浓度偏高的项目,双细胞检测尤其重要。
4. 从 scRNA-seq矩阵到可解释结果,通常要经历4步
4.1 标准化
原始 scRNA-seq矩阵的不同细胞测序深度不同,不能直接比较。标准化的目的,是让表达值更可比。常见做法包括按总 UMI 归一化,再进行对数转换。
标准化不是为了美化数据,而是为了降低测序深度差异带来的偏差。 这一步直接影响后续聚类、可视化和差异表达分析。
4.2 高变基因筛选
并不是所有基因都适合用于下游分析。高变基因能更好地反映细胞间差异。它们通常被用于降维和聚类。
如果把大量稳定表达基因一起纳入,信号会被稀释。因此,高变基因筛选是从 scRNA-seq矩阵中提取有效信息的重要步骤。
4.3 降维和聚类
PCA、UMAP、t-SNE 等方法常用于把高维矩阵压缩到低维空间。这样可以更直观地看到细胞群体结构。随后再进行聚类,寻找可能的细胞亚群。
但要注意,降维图只是可视化结果,不是生物学终点。聚类结果必须回到 marker 基因、已知文献和实验背景中验证。
4.4 细胞注释
注释是把数据簇对应到具体细胞类型的过程。常结合经典标记基因、数据库和参考图谱完成。对于临床样本或疾病队列,注释尤其要谨慎。
同一个簇不一定是一个纯净细胞群。可能存在状态转换、激活谱变化,甚至混入少量污染细胞。因此,scRNA-seq矩阵分析的最终目标不是“分出更多簇”,而是得到可解释、可复现的生物学结论。
5. 高质量 scRNA-seq矩阵分析,离不开规范化工作流
5.1 先质控,再整合,再解释
一个稳妥的顺序通常是,先做细胞和基因质控,再做标准化和批次处理,最后进入降维、聚类和注释。顺序错了,后面的结果往往要返工。
在团队协作中,最好保留完整参数记录,包括过滤阈值、归一化方法、整合策略和版本信息。这不仅关系到结果复现,也关系到论文审稿和后续投稿。
5.2 让数据分析服务于研究问题
不同课题对 scRNA-seq矩阵的要求不同。肿瘤研究更关注异质性和微环境,免疫研究更关注状态谱系,发育研究更关注轨迹变化。分析策略必须围绕研究问题设计,而不是机械套模板。
如果目标是发现罕见细胞,就要重视深度和稀疏性处理。如果目标是比较患者与对照,就要更关注批次平衡和样本结构。研究问题不同,矩阵解读方式也不同。
5.3 借助专业工具减少重复劳动
对于需要高频处理单细胞项目的团队,使用成熟平台能显著提升效率。解螺旋品牌提供的单细胞数据分析相关产品和服务,可以帮助研究者更系统地完成 scRNA-seq矩阵质控、标准化、整合和注释,减少手工试错成本。 这对临床研究、课题组和转化项目都更友好。
在实际工作中,规范流程加上可靠工具,往往比“临时拼接多个软件”更稳定,也更利于结果复现。
总结Conclusion
scRNA-seq矩阵是单细胞研究的核心起点。读懂它,关键在于抓住5件事:矩阵结构、质量指标、稀疏性与批次效应、标准化分析流程,以及面向研究问题的注释策略。只有先把矩阵处理正确,后面的生物学解释才可信。
如果你的课题正在处理单细胞数据,但卡在 scRNA-seq矩阵 的质控、整合或注释环节,建议优先使用更成熟的方案。解螺旋品牌能帮助你把复杂流程标准化,提升分析效率与结果可信度。
- 引言Introduction
- 1. scRNA-seq矩阵到底是什么
- 2. 解读 scRNA-seq矩阵前,先看3个核心质量指标
- 3. scRNA-seq矩阵最容易忽视的3类问题
- 4. 从 scRNA-seq矩阵到可解释结果,通常要经历4步
- 5. 高质量 scRNA-seq矩阵分析,离不开规范化工作流
- 总结Conclusion