引言Introduction
测序reads是二代测序和单细胞分析中最基础的结果单位。很多初学者看得到reads数,却不知道它代表什么、能否直接比较、以及如何影响表达定量。理解测序reads,是读懂测序报告和后续分析的第一步。
1. 测序reads到底是什么
1.1 从“片段序列”理解reads
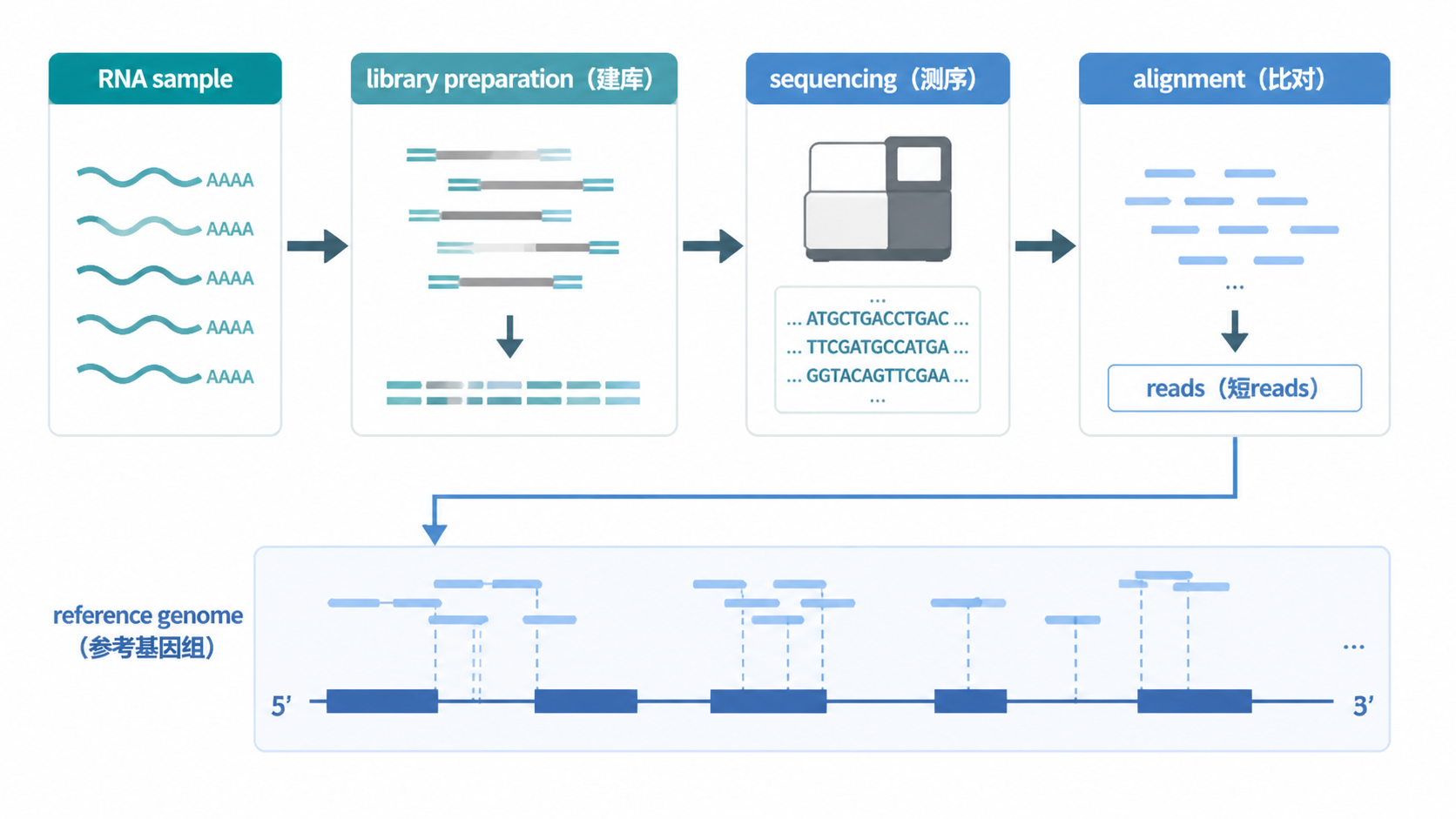
测序reads,本质上就是测序仪读出来的一段段短序列。 在二代测序中,样本先被打断成很多小片段,再进行测序。每一段被读取到的序列,都可以看作一个read。
在RNA测序里,reads来源于cDNA片段。RNA先去除rRNA,再打断mRNA,逆转录成cDNA,最后进入测序流程。对研究者来说,reads不是抽象概念,而是后续做比对、定量、差异分析的最小数据单元。
1.2 reads和表达量不是同一个概念
reads多,不等于真实表达一定高。 因为reads数量受多个因素影响,包括基因长度、测序深度、文库质量和比对效率。
例如,同一样本中,较长的基因往往会被打断成更多片段,产生更多reads。若不做标准化,直接比较不同基因的reads数量,容易得出偏差结论。
2. reads是怎么产生的
2.1 二代测序的核心原理
二代测序的核心是“边合成边测序”。在DNA合成过程中,系统通过荧光信号识别每次加入的碱基,从而得到序列信息。这个过程依赖大规模平行测序,所以能同时读取大量片段。
简而言之,reads是测序仪把样本片段“读出来”的结果。 这也是NGS高通量的基础。
2.2 从RNA到reads的完整路径
在RNA-Seq中,样本通常要经历以下步骤:
- 去除rRNA。
- 打断mRNA。
- 随机引物逆转录成第一条cDNA。
- 合成第二条DNA。
- 加接头,完成建库。
- 扩增并上机测序。
- 生成原始reads。
- 比对到参考基因组。
- 进行表达定量。
这个流程决定了reads并不是“天然存在”的,而是实验和计算共同生成的结果。
3. 为什么reads不能直接拿来比较
3.1 基因长度会影响reads数量
同一个样本中,不同基因的reads数不能直接比较。原因很简单。基因越长,随机打断后得到的片段通常越多,被测到的概率也更高。
这意味着,一个reads更多的基因,不一定就是更高表达,只可能是它更长。
3.2 测序深度也会影响reads数量
同一个基因在不同样本中的reads数,也不能直接比较。因为样本间测序深度不同。深度越深,被随机抽中的机会越大,reads就可能更多。
因此,分析时通常需要做标准化处理。常见思路是同时校正基因长度和测序深度,再比较样本间表达差异。不校正就比较,结论往往不可靠。
3.3 文库质量也会改变reads分布
如果样本RNA完整性较差,建库效果会受影响。上游知识库提到,RIN值是判断RNA完整性的关键指标,通常RIN大于9,说明RNA降解较少,更适合建库测序。
样本质量决定reads质量。 这一点在临床样本、石蜡包埋样本和低起始量样本中尤其重要。
4. 读懂测序reads要看哪些核心指标
4.1 reads数只是第一步
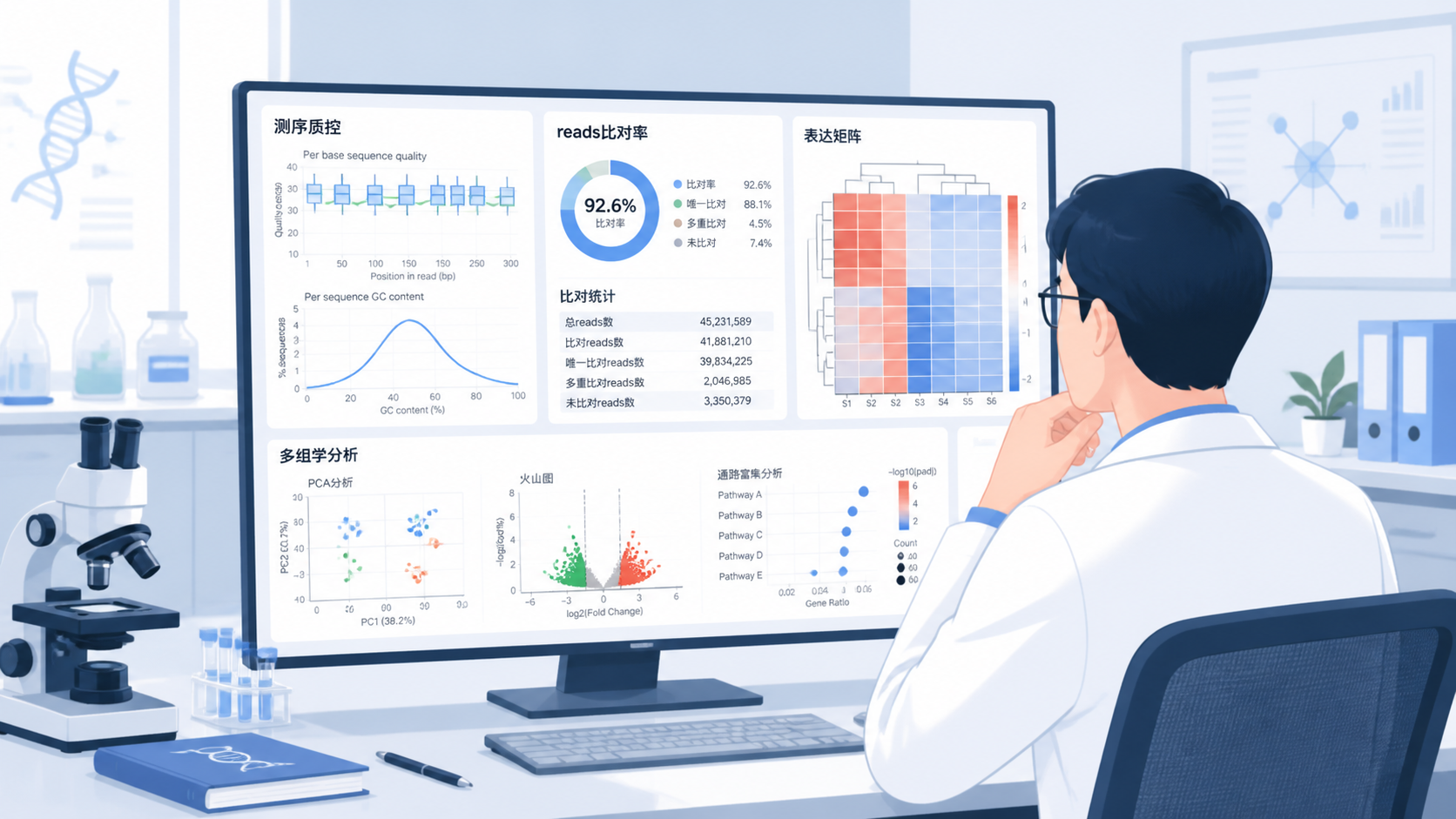
很多人只看总reads数,但这远远不够。真正有分析价值的,是reads后的多个质量指标。
常见关注点包括:
- 原始reads数。
- 比对率。
- 唯一比对reads比例。
- 重复序列比例。
- 覆盖深度。
- 每个基因的read count。
总reads高,不代表结果一定好。 如果比对率低,或者大量reads落在低复杂度区域,数据仍可能不理想。
4.2 read count和表达定量的关系
在表达分析中,read count是最常见的定量结果。它表示某个基因被比对到的reads数量。随后还需要结合标准化方法,得到可比较的表达矩阵。
对于科研人员来说,这一步非常关键。因为后续差异表达、富集分析、细胞亚群比较,几乎都建立在read count或其标准化结果之上。
4.3 比对结果决定reads能否被利用
测序得到的短序列,需要比对到参考基因组或参考转录组。常用软件包括Hisat2和STAR。只有成功比对的reads,才真正进入后续定量和生物学解释。
如果比对率偏低,要优先排查参考基因组版本、样本污染、RNA质量和建库过程,而不是直接进入下游分析。
5. 测序reads在实际研究中怎么用
5.1 读取研究问题的入口
reads不是终点,而是入口。它帮助研究者回答三个基础问题:
- 样本里有哪些转录本被测到。
- 这些转录本大致有多少。
- 样本间表达差异是否存在。
对于单细胞测序,reads进一步支撑细胞类型注释、亚群识别和稀有细胞检测。对于bulk RNA-Seq,reads常用于差异表达和通路分析。
5.2 什么时候更应该关注reads质量
以下几类场景要特别关注:
- 低丰度转录本检测。
- 稀有细胞群识别。
- 临床样本RNA质量不稳定。
- 石蜡包埋组织分析。
- 需要发现融合基因、SNP、INDEL等变异。
reads质量越稳定,结果越可信。 这也是高质量测序服务和规范化分析的重要价值。
5.3 测序与芯片的思路差异
上游知识库提到,RNA-Seq可检测新转录本、基因融合和多种变异,动态范围更大;而芯片在定量上更成熟,流程更简单。若研究目标是探索未知变化,测序更合适。若更强调成熟定量和简单流程,芯片也有优势。
这说明reads的价值,不只是“有多少”,更在于它能支持什么研究目标。
6. 研究者该如何正确看待测序reads
6.1 先看质量,再看数量
先看比对率、RIN值、文库质量,再看reads数量。 这是更专业的判断顺序。只盯着reads总数,很容易忽略前处理问题。
6.2 先做标准化,再做比较
无论是不同基因之间,还是不同样本之间,都要先校正长度和深度差异。否则reads高低的比较没有统计学意义。
6.3 结合研究目的解释结果
如果目的是找差异表达,就关注标准化后的表达矩阵。
如果目的是发现新转录本或融合事件,就要重视reads覆盖和比对模式。
如果目的是单细胞分群,就要关注每个细胞的有效reads和捕获效率。
reads的解读必须回到研究问题本身。 这才符合E-E-A-T意义上的专业分析。
总结Conclusion
测序reads是测序仪读取到的短序列片段,也是RNA-Seq和单细胞分析中最基础的数据单元。它能反映样本中的转录信息,但不能脱离基因长度、测序深度和样本质量单独解读 。真正专业的做法,是先看质控,再看比对,再做标准化定量。
如果你正在做测序设计、结果解读或论文写作,建议把reads理解成“起点”而不是“结论”。想更高效地完成测序数据分析,可以关注解螺旋 ,获取更适合科研场景的测序分析支持与品牌服务。
- 引言Introduction
- 1. 测序reads到底是什么
- 2. reads是怎么产生的
- 3. 为什么reads不能直接拿来比较
- 4. 读懂测序reads要看哪些核心指标
- 5. 测序reads在实际研究中怎么用
- 6. 研究者该如何正确看待测序reads
- 总结Conclusion