引言Introduction
无代码TCGA分析正在降低生信门槛,但很多医学生、医生和科研人员仍卡在“数据从哪来、怎么清洗、怎么出图”这一步。其实,只要把流程拆成5个关键步骤,就能把TCGA数据分析做得更稳、更快。
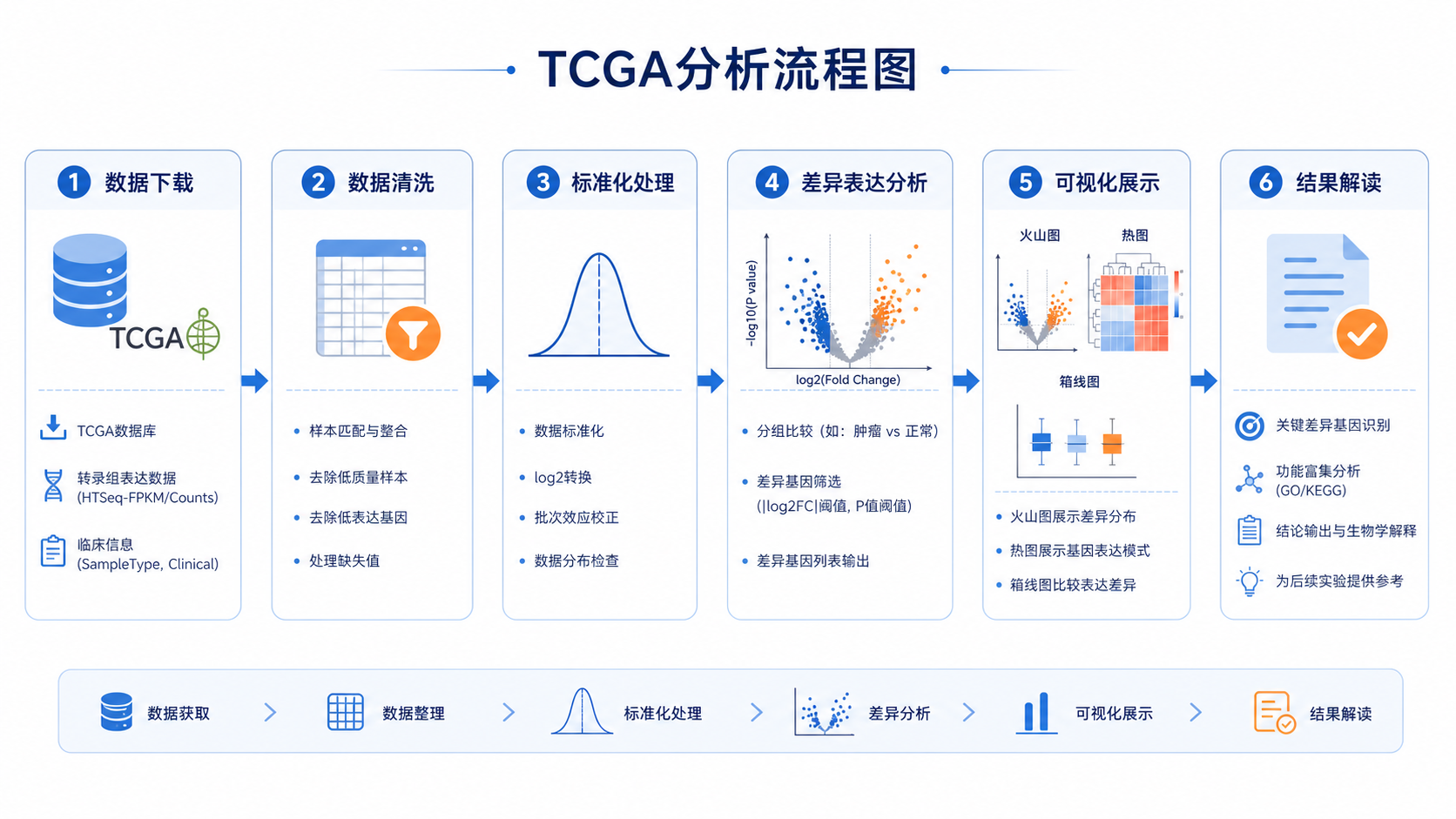
1. 先搭好项目结构,再谈分析
1.1 明确文件夹分工
无代码TCGA分析并不是“不写代码”,而是把复杂流程标准化。第一步要先理清项目结构。常见做法是把下载数据、注释文件、分析结果、图表输出分开存放。这样后续查找、复现和汇报都会更高效。
在课程示例中,核心代码通常集中在一个主目录下,包含自定义函数、差异分析代码、GO/KEGG分析代码,以及TCGA数据下载脚本。先把目录整理清楚,能直接减少后续80%的混乱。
1.2 先确认分析对象
TCGA数据类型很多。RNA-seq、临床信息、metadata、样本质量注释文件,都可能参与分析。真正开始前,要先明确研究问题。
例如,若目标是肝癌差异分析,就要优先确认肿瘤样本、正常样本、配对样本是否齐全。之后再决定是否纳入临床结局、预后模型或外部验证。
1.3 先做可复现准备
无代码TCGA分析要想用于科研发表,必须强调可复现性。至少要保留以下内容。
- 数据来源与版本
- 样本筛选规则
- 关键参数
- 结果输出路径
- 代码或流程记录
没有流程记录,结果就很难复现。
2. 数据下载后,先做清洗和样本质控
2.1 先整理metadata
TCGA分析最容易出问题的,不是差异分析本身,而是前期样本整理。metadata文件通常包含文件名、样本全名、TCGA ID、病人编号、组织类型等信息。需要先把这些字段提取出来,并统一成可分析的数据表。
在实际流程里,常会新增分组信息。比如,11代表正常,其余代表肿瘤。这样后续分组统计更直观,也更便于差异分析。
2.2 样本质量过滤不能省
样本质控是关键步骤。课程示例中,会读取样本质量注释文件,再逐步筛掉不合适的样本。最终共过滤掉23个样本。这个数字说明,原始TCGA数据并不总是“拿来就能用”。
常见的处理原则包括。
- 检查样本注释是否完整。
- 核对barcode是否匹配表达矩阵。
- 剔除质量差或注释不一致的样本。
- 保留能明确归类到肿瘤或正常的样本。
样本筛错,比模型选错更致命。
2.3 配对样本优先保留
如果研究设计允许,配对样本更适合做差异分析。比如同一患者的肿瘤组织和正常组织同时存在时,统计效应更稳。课程中整理后得到44个病人的88个组织样本,这类设计能显著减少个体差异干扰。
对于医学生和科研人员来说,记住一个原则就够了。配对样本优先,非配对样本次之。
3. 做差异分析前,先把表达矩阵处理干净
3.1 过滤低表达基因
原始RNA-seq数据里,低表达基因很多。这些基因噪音大,容易干扰差异结果。常见做法是先筛掉表达量为0或接近0的基因,只保留有实际信号的基因。
课程中对mRNA表达数据进行了筛选,提取protein coding基因,并进一步保留表达量大于0的基因。这一步看似简单,却直接影响后面火山图和热图的可信度。
3.2 标准化后再比较
不同样本测序深度不一样,原始count不能直接比。常用的做法是借助DESeq2的VST函数进行标准化。这样可以让样本间的表达分布更接近,减少技术偏差。
标准化后,还要做质量评估。常见检查包括。
- 核形图
- 密度图
- PCA图
如果PCA图显示分组趋势明显,说明数据结构相对合理。若样本明显离群,就要回到前面重新检查质控和分组。
3.3 无代码流程也要理解统计逻辑
即使使用可视化平台或封装流程,也不能跳过统计原理。差异分析本质上仍是比较两组表达差异,并结合logFC和P值判断显著性。课程中使用DESeq2进行差异表达基因筛选,并根据logFC和P值给出上调、下调或无显著差异的分类。
只有理解统计逻辑,才能避免“只会点按钮,不会解释结果”。
4. 把差异结果转成能发表、能解读的图
4.1 火山图负责快速定位重点基因
差异分析完成后,第一类核心图通常是火山图。它能同时展示logFC和显著性,帮助快速定位上调和下调基因。对于大规模TCGA数据,火山图是最直观的结果入口。
在课程提纲中,使用ggplot2和ggrepel绘制火山图,这是科研写作中最常见、也最容易被审稿人接受的展示方式之一。
4.2 热图帮助看表达模式
热图更适合展示重点基因在样本间的表达模式。例如课程中提取了15个有氧呼吸相关差异基因,并用pheatmap绘制表达矩阵。
热图的价值在于,它不仅告诉你“谁差异显著”,还告诉你“这些基因在样本中是否形成一致模式”。
4.3 进一步做功能解释
如果只停留在差异基因列表,结果往往不够完整。后续通常要做GO和KEGG富集分析,解释这些基因可能参与哪些生物过程、分子功能和通路。对于肝癌等实体瘤研究,这一步可以把“数据差异”转换成“机制假设”。
从差异基因到通路解释,才算完成一轮有价值的TCGA分析。
5. 把结果推进到临床问题,才真正有价值
5.1 从差异基因走向预后模型
无代码TCGA分析的最终目的,不只是画图,而是服务临床问题。课程的完整套路中,会将TCGA差异分析结果与临床生存数据结合,进一步做单因素、多因素和相关性分析,建立预后模型。
对于科研人员来说,这一步最重要的意义在于,把分子层面的结果转化为临床可用的信息。比如高危与低危分层、患者生存预测、个体化治疗参考。
5.2 用外部队列验证
模型如果只在TCGA内部成立,证据还不够强。课程提纲中还提到使用ICGC数据库进行外部验证。外部验证的作用非常明确,检验模型是否具有泛化能力。
没有外部验证,很多结论只能算“初步发现”。
5.3 用标准化产品提升效率
如果你希望少走弯路,减少手工整理和重复试错,建议直接使用成熟的流程化工具。像解螺旋这类品牌化产品,可以把TCGA下载、样本整理、差异分析、可视化和预后分析串成更清晰的工作流。
对于医学生和科研人员来说,这意味着更少的手动失误,更快的出图速度,以及更稳定的结果复现。把复杂流程交给标准化工具,才能把时间留给科学问题本身。
总结Conclusion
无代码TCGA分析的核心,不是省略步骤,而是把复杂流程标准化。你需要依次完成项目结构梳理、数据清洗与质控、表达矩阵标准化、差异结果可视化,以及临床转化验证。只要这5步做扎实,TCGA分析就能更稳、更快,也更适合科研发表。

如果你希望进一步提升效率,减少重复劳动,可以结合解螺旋 的标准化流程工具,把无代码TCGA分析做得更规范、更高效。
- 引言Introduction
- 1. 先搭好项目结构,再谈分析
- 2. 数据下载后,先做清洗和样本质控
- 3. 做差异分析前,先把表达矩阵处理干净
- 4. 把差异结果转成能发表、能解读的图
- 5. 把结果推进到临床问题,才真正有价值
- 总结Conclusion