






引言Introduction
TCGA样本数据筛选做不好,后续差异分析、分型和预后模型都会被批次效应、样本类型错误和低质量数据拉偏。对医学生、医生和科研人员来说,真正难点不是“下载数据”,而是先把样本筛干净 。
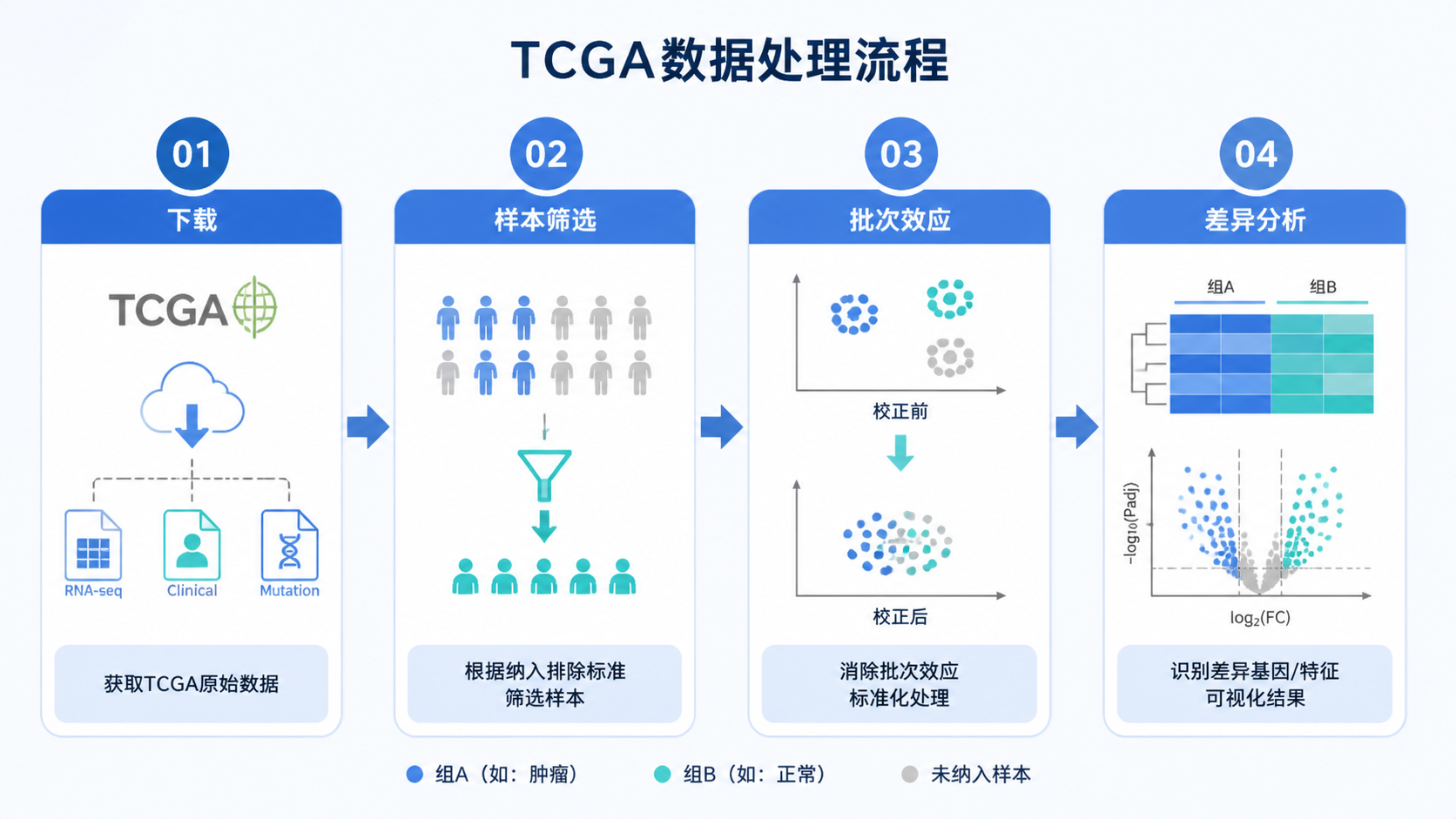
1.TCGA样本数据筛选的第一步,是先看Metadata和barcode
1.1 Metadata决定你能不能准确匹配样本
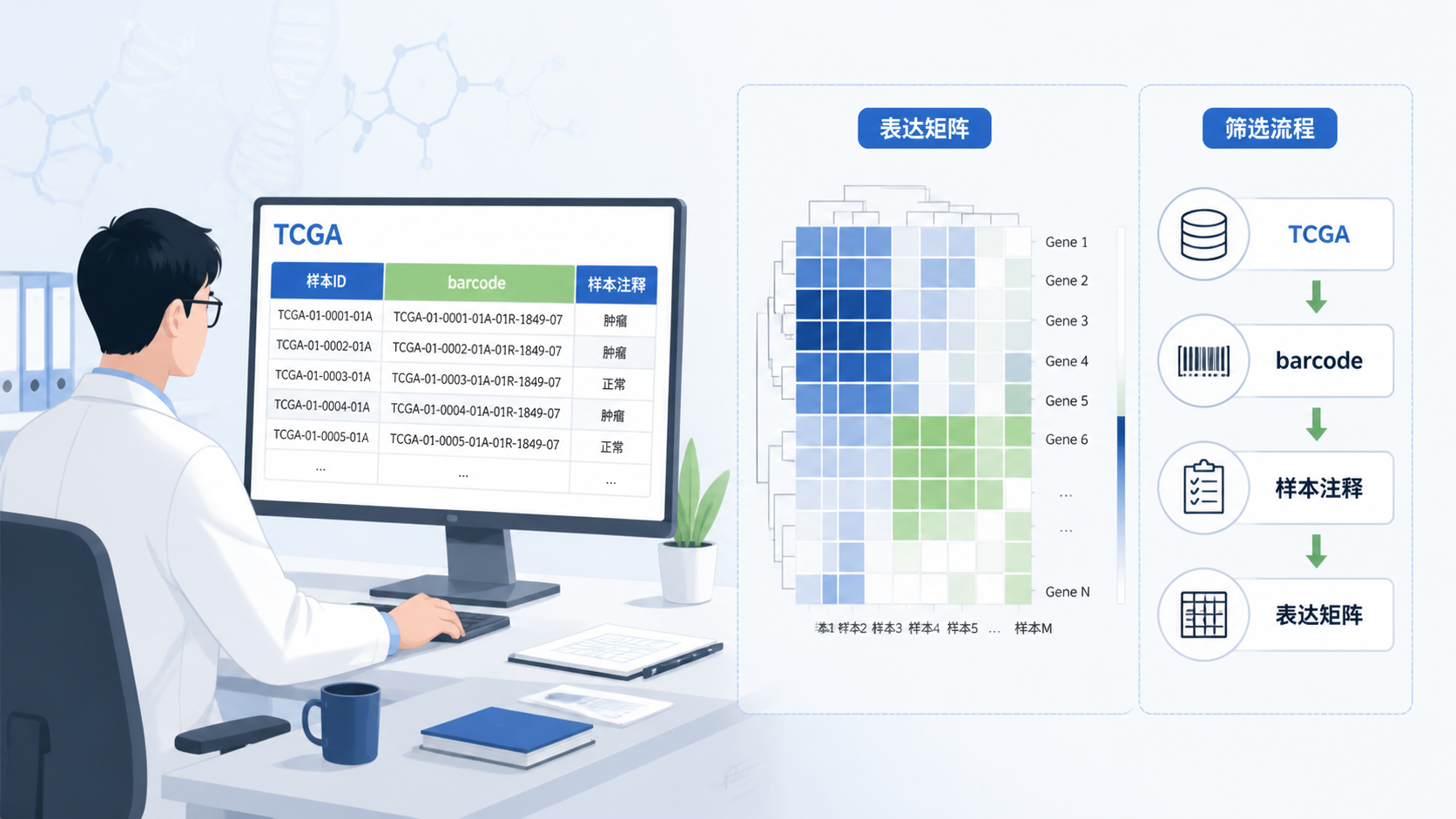
TCGA下载页面里的Metadata,通常以JSON格式导出。它包含样本名称、样本ID、组织类型等关键信息。原始数据文件名并不等于样本ID ,如果不先做对应关系整理,后面很容易把表达矩阵和临床信息接错。
实际操作中,建议先用Metadata提取文件名和sample ID的对应关系,再结合manifest文件整理样本注释。这样做的价值很直接,能把“文件属于谁”这件事先定下来,避免后续分析建立在错误样本上。
1.2 barcode是样本分组的核心依据
TCGA barcode是每个样本的唯一识别码,编码结构固定,信息非常丰富。其中第14、15位字符最关键。01代表原发实体肿瘤,11代表实体正常组织 。在TCGA样本数据筛选中,这两位字符常被直接用于判定肿瘤组和正常组。
例如,在RNA测序数据里,常见的组织类型编码包括01、02、03、05、06、07和11。只有11是正常组织,其余多为肿瘤相关样本 。因此,做差异分析前,先用barcode提取分组信息,是最基础也最重要的一步。
2.TCGA样本数据筛选的第二步,是识别并处理批次效应
2.1 不能只看分组,还要看TSS、plate和center
很多人做TCGA样本数据筛选,只关注肿瘤和正常,却忽略了批次信息。实际上,TCGA barcode里还能读出TSS、plate和center。它们分别反映来源中心、测序板和分析中心,都会带来批次效应。
课程内容强调,批次效应至少有两类常见来源,TSS编码和测序板信息 。如果不同样本来自不同中心,或在不同板上处理,表达差异可能部分来自技术偏差,而不是生物学差异。
2.2 合并GTEx和TCGA时,批次问题更要谨慎
如果TCGA样本不足,很多研究会把GTEx正常样本合并进来。这个思路本身合理,但前提是要使用统一重新分析的数据。不建议把两个数据库各自下载后直接拼接表达矩阵 ,因为处理流程不同,技术差异会被放大。
更稳妥的做法,是从UCSC等统一重计算平台获取数据,再进行合并。合并后仍可能存在批次效应,可结合RUVSeq或SVA处理。对TCGA样本数据筛选来说,这一步不是“附加项”,而是决定结果是否可信的关键控制点。
3.TCGA样本数据筛选的第三步,是严格核查样本注释
3.1 annotation表比表面分组更重要
TCGA官网的annotation信息,不能只看样本名,还要看病例注释。因为有些样本虽然来源于某个器官,但病理诊断可能并不属于你研究的疾病类型。这类样本如果不剔除,会直接污染研究队列 。
课程中提到肝癌数据的例子。某些样本在肝脏来源上看似符合,但病理上实际可能是胆管癌。若你研究的是LIHC,这类样本应剔除;若研究CHOL,则可能应保留。样本来源不等于疾病归属 ,这是TCGA样本数据筛选里最容易忽视的误区。
3.2 先筛注释,再筛表达矩阵
实操顺序建议是先读注释表,再回到表达矩阵筛样本。可以先下载TCGA pan-cancer相关的样本质量注释文件,逐个检查是否存在不适合纳入分析的条目。对于有明确说明的样本,优先按注释处理;对没有注释的样本,再结合barcode和临床信息判断。
样本筛选不是简单删几列,而是建立研究队列的过程。 这一步做得越严谨,后面差异分析的解释力越强,审稿时也越容易通过质疑。
4.TCGA样本数据筛选的第四步,是只保留适合统计分析的样本
4.1 肿瘤和正常分组要明确
在TCGA RNA-seq数据中,最常用的分组逻辑就是第14、15位字符。课程中明确提到,做差异分析时通常提取01和11样本。01为肿瘤,11为正常 ,这是最清晰、最可复用的分组方式。
如果你的研究是配对设计,还要进一步检查是否同一患者同时拥有肿瘤和正常组织。课程提到,可以按patient ID统计,筛出同时具备01和11样本的病例。这样得到的配对样本,统计解释更强,也更适合配对差异分析。
4.2 过滤掉不合适的样本数量,宁可少一点,也不要混一点
课程案例中,对肝癌样本进行了样本质量过滤,最终剔除了一批不合适样本。这个做法体现了一个原则:宁可样本少,也不要把错误样本混进来 。因为错误样本带来的偏差,往往比样本量减少造成的统计损失更严重。
对医学生和科研人员来说,队列构建的底线是,样本归属明确、分组逻辑一致、注释信息完整。如果这三点做不到,后续的火山图、热图和通路富集都可能失真。
5.TCGA样本数据筛选的第五步,是做基因层面的过滤
5.1 先去掉不表达和低表达基因
样本筛完之后,还要做基因过滤。RNA-seq差异分析前,通常要去掉全为0或低表达基因。过滤低表达基因的目的,是提高差异分析的敏感性和准确度 。这一步看似简单,但对结果稳定性影响很大。
课程中给出多个常用策略。最基本的是去除表达量全为0的基因。更进一步,可以保留至少一半样本中表达量大于0的基因,或者保留中位数大于0的基因。没有统一最优标准,应该结合样本量和分组设计判断。
5.2 平均值过滤要谨慎
有些人会用平均值做过滤,但要注意,平均值可能掩盖组间极端差异。比如某个基因在一组完全不表达,在另一组高表达,平均值有时会把它误删。因此,平均值过滤要设得更保守,不能一刀切。
如果你分析的是TCGA样本数据筛选后的表达矩阵,建议优先采用“是否表达”“中位数是否大于0”“至少多少比例样本表达”这类更稳健的规则。这样更符合差异分析的统计逻辑。
6.把筛选流程做成标准化步骤,才适合长期复用
6.1 一个更稳妥的TCGA样本数据筛选顺序
如果你想把流程固定下来,可以按下面顺序执行:
- 下载Metadata和manifest,建立文件名与样本ID对应关系。
- 读取barcode,提取组织类型和样本分组。
- 核查annotation,剔除病理归类不一致样本。
- 检查TSS、plate、center等批次信息。
- 过滤掉低质量样本和不适合纳入分析的样本。
- 再进行基因过滤,保留适合差异分析的表达矩阵。
这个顺序的优势是清晰、可追溯,也方便后续在R或其他生信流程中复现。
6.2 对科研结果最有帮助的,不是样本最多,而是样本最干净
很多项目失败,不是因为数据太少,而是因为筛选不严。TCGA样本数据筛选做得好,后续无论是差异表达、分型分析,还是预后模型,都更容易得到稳定结果。真正可靠的分析,往往始于严格的样本清洗。
如果你希望把TCGA样本数据筛选、GTEx合并、ID转换和过滤流程一次性规范化,借助解螺旋的标准化课程和数据处理思路,会比零散试错更高效,也更适合科研项目落地。
总结Conclusion
TCGA样本数据筛选的核心,不是简单删样本,而是围绕Metadata、barcode、annotation、批次效应和基因过滤,建立一套可复现的标准流程。只要这5个要点处理到位,后续差异分析的可信度会明显提高。
如果你正在做TCGA相关研究,建议把样本筛选当作正式方法学步骤来写。 这样不仅更严谨,也更符合E-E-A-T要求。需要更系统的TCGA处理方案时,可以进一步参考解螺旋的标准化生信课程与工具,减少试错,提高分析效率。
- 引言Introduction
- 1.TCGA样本数据筛选的第一步,是先看Metadata和barcode
- 2.TCGA样本数据筛选的第二步,是识别并处理批次效应
- 3.TCGA样本数据筛选的第三步,是严格核查样本注释
- 4.TCGA样本数据筛选的第四步,是只保留适合统计分析的样本
- 5.TCGA样本数据筛选的第五步,是做基因层面的过滤
- 6.把筛选流程做成标准化步骤,才适合长期复用
- 总结Conclusion



