引言Introduction
单细胞数据质控决定后续分析是否可信。很多项目不是“不会分析”,而是前期低质量细胞混入太多 ,导致聚类偏移、差异基因失真,甚至结论错误。本文围绕单细胞数据质控 的5大关键指标,帮助医学生、医生和科研人员快速建立全局判断框架。
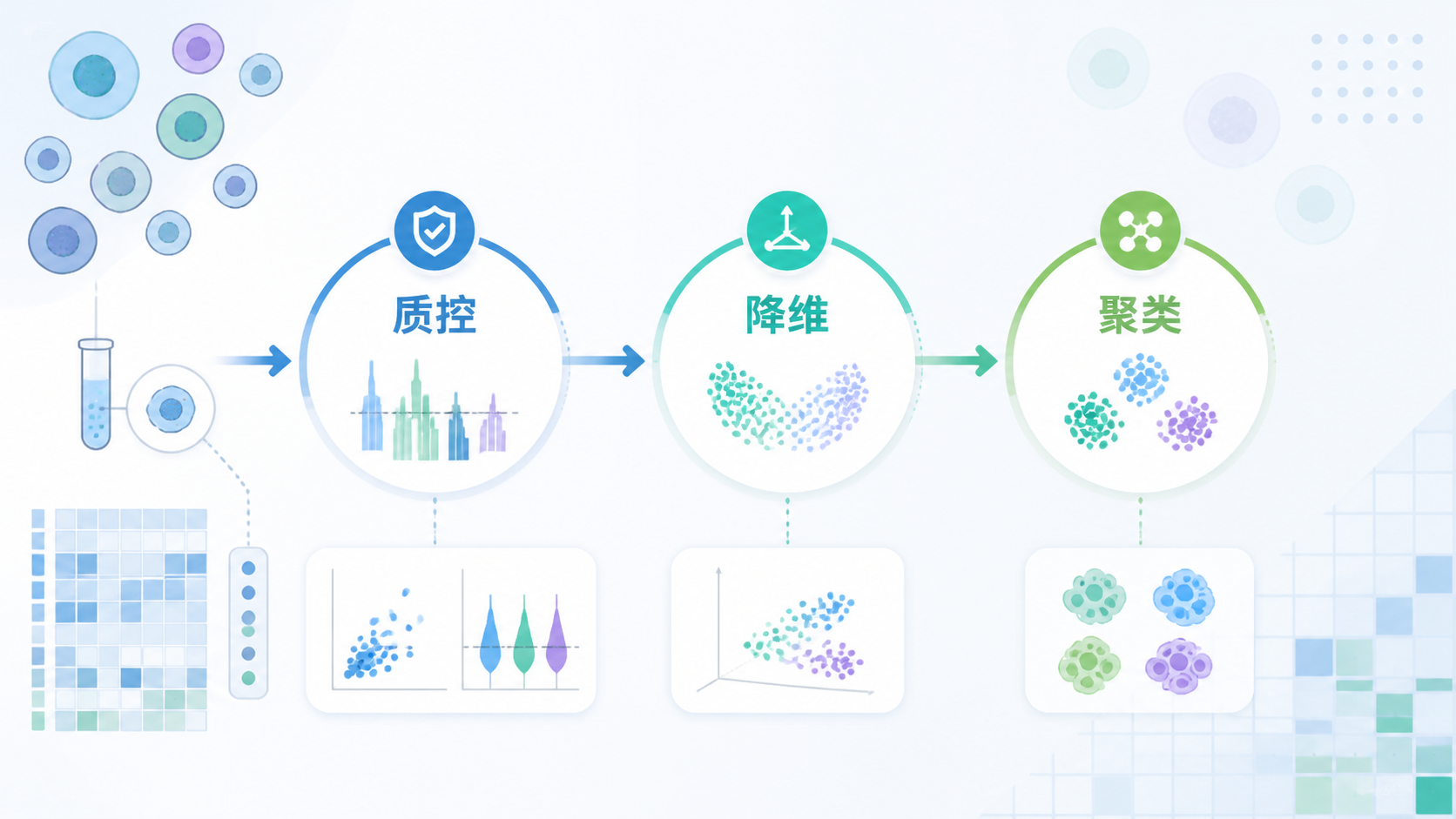
1. 为什么单细胞数据质控是第一步
1.1 质控的目标不是“删数据”,而是保留可信细胞
单细胞实验常见问题有两类。第一类是低质量细胞,表现为测到的基因少、转录本少,或比对率异常低。第二类是混入异常细胞,例如双细胞或多细胞油滴,会直接扭曲表达矩阵。
单细胞数据质控的核心目标,是尽早排除会污染后续分析的细胞。
这一步做得好,后面的降维、聚类和差异分析才有意义。
1.2 质控结果会直接影响下游分析
上游课程案例中,原始数据为2,579个细胞,经过基础过滤后保留2,210个细胞,保留率约86%。这说明质控并不是少删越好,而是要按数据分布设置合理阈值。
如果低质量细胞保留过多,常见后果包括:
- PCA、t-SNE或UMAP图出现“噪音尾巴”。
- 聚类边界模糊,亚群不稳定。
- marker基因表达被稀释。
- 差异分析出现假阳性或假阴性。
2. 关键指标一:每个细胞检测到的基因数
2.1 基因数反映细胞信息完整度
在单细胞数据质控中,基因数是最直观的指标之一。一个细胞检测到的基因越少,说明捕获到的转录信息越有限,往往提示细胞质量差、RNA降解,或者建库效率不足。
课程中给出的经验阈值是2,000个基因 。
对于该胚胎数据,这是合理的,因为胚胎细胞和干性较强的生殖细胞本身转录活性高。
2.2 阈值不能照搬,要结合样本类型
2,000个基因不是通用标准。
不同体系差异很大。
例如:
- 胚胎组织、干性细胞,通常基因数更高。
- 成体组织、低RNA含量细胞,阈值可能需要下调。
- 10x或Drop-seq数据,某些场景下200到500个基因也可能用于初筛。
因此,判断时要看整体分布,而不是只盯着单个数值。
单细胞数据质控的关键,是依据样本背景设阈值。
3. 关键指标二:每个细胞的转录本数或UMI数
3.1 转录本数反映测序深度和捕获效率
转录本数,或UMI count,代表一个细胞被捕获到的分子总量。它既能反映测序深度,也能间接反映建库质量。
课程案例中,设置了10万转录本数 作为阈值。
原因是该数据整体质量很好,且研究对象是胚胎生殖细胞,表达量本来就高。
3.2 过低和过高都要警惕
转录本数过低,常见于:
- 细胞破裂。
- RNA降解。
- 捕获失败。
转录本数异常过高,则要怀疑:
- 不是单细胞,而是双细胞或多细胞。
- 存在异常高背景信号。
- 文库复杂度异常。
单细胞数据质控不是只看“低值剔除”,高异常值同样重要。
4. 关键指标三:比对率或Mapping rate
4.1 比对率反映测序内容是否可靠
比对率是指测序reads中成功比对到参考基因组的比例。
如果一个细胞的reads大多比不到参考序列,说明它的文库可能存在问题,或者样本质量本身就差。
这类细胞继续保留,会把技术噪音带入后续分析。
比对率低,是单细胞数据质控中非常重要的报警信号。
4.2 适合用于排查系统性问题
比对率异常时,往往不只是单个细胞的问题,还可能提示:
- 组织解离不充分。
- RNA质量差。
- 文库构建失败。
- 参考基因组或注释文件不匹配。
因此,比对率不仅用于过滤细胞,也可用于回头检查实验流程。
这是E-E-A-T意义上的“经验判断”:质控既看结果,也看原因。
5. 关键指标四:总reads数和异常高值细胞
5.1 总reads数能提示非单细胞风险
课程中提到,单细胞总reads数也值得关注。
如果某个细胞的reads总数异常偏高,不能简单认为它“质量很好”。它也可能是双细胞甚至多细胞混合进入同一油滴。
这类细胞在表达矩阵中会表现为:
- 基因数偏高。
- UMI偏高。
- marker混杂。
- 聚类位置异常。
5.2 为什么高reads细胞会污染分析
高reads细胞会让聚类算法误判相似性。
比如两个原本独立的细胞类型,被一个混合细胞“桥接”,就可能在图上形成错误过渡。
因此,单细胞数据质控要同时排除低值和高异常值。
只删低质量细胞,远远不够。
6. 关键指标五:高变基因或高抑制性基因的选择
6.1 不是所有基因都适合用于后续分析
单细胞转录组通常有2万多个基因,但真正能区分细胞类型的只是其中一部分。
大多数基因在不同细胞之间表达差异不明显,另一些则可能受测序技术影响,噪音较大。
因此,后续分析通常要筛选能体现细胞差异的基因。
课程中将其描述为高抑制性基因 ,本质上是挑出更能代表不同细胞类型特征的信号。
6.2 这一步关系到聚类和注释质量
如果特征基因筛选不合理,常见问题包括:
- 细胞亚群分不开。
- 聚类结果不稳定。
- marker基因信号不突出。
- 注释偏差增大。
从分析流程看,单细胞数据质控不止是过滤细胞,也包括筛选用于建模的特征基因。
这一步会显著影响后续降维和聚类的可解释性。
7. 实战中如何建立一套可复用的质控流程
7.1 先看分布,再定阈值
建议按以下顺序处理:
- 查看每个细胞的基因数分布。
- 查看UMI或转录本数分布。
- 查看比对率分布。
- 排查异常高reads细胞。
- 再决定是否进一步保留高变基因。
不要先设固定阈值,再强行套到所有项目。
7.2 阈值要结合组织来源和实验平台
胚胎样本、成体样本、肿瘤样本、免疫细胞样本,质控阈值都可能不同。
10x Genomics、Drop-seq、Smart-seq2的信号特征也不同。
因此,真正规范的做法是:
- 先看全局分布。
- 再结合样本生物学背景。
- 最后确定过滤策略。
这也是高质量单细胞数据质控的标准思路。
7.3 建议把质控和结果回看结合起来
质控不应是一次性动作。
完成聚类后,还要回看:
- 是否存在明显低质量簇。
- 是否某个簇以高线粒体或低基因为特征。
- 是否存在疑似双细胞群。
质控是贯穿全流程的,而不是只发生在最开始。
总结Conclusion
单细胞数据质控决定了后续分析的可信度。真正需要关注的,不只是“删掉低质量细胞”,还包括基因数、转录本数、比对率、总reads数和特征基因筛选这5个关键环节。
先看分布,再定阈值,再结合样本背景调整策略 ,这是更稳妥的分析逻辑。
如果你希望把这些步骤标准化,减少重复摸索,可以借助解螺旋的单细胞分析内容与工具支持,把质控、筛选和下游分析串成一条清晰流程。这样,单细胞数据质控就不再是经验猜测,而是可复用、可追踪、可解释的规范流程。
- 引言Introduction
- 1. 为什么单细胞数据质控是第一步
- 2. 关键指标一:每个细胞检测到的基因数
- 3. 关键指标二:每个细胞的转录本数或UMI数
- 4. 关键指标三:比对率或Mapping rate
- 5. 关键指标四:总reads数和异常高值细胞
- 6. 关键指标五:高变基因或高抑制性基因的选择
- 7. 实战中如何建立一套可复用的质控流程
- 总结Conclusion